In suspected cases, an evaluation with history (including family history), physical examination, ECG, and echocardiography should be performed. The latter establishes the diagnosis. Asymptomatic patients are usually diagnosed at the time of routine heart examination or family screening.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
Patients are most commonly diagnosed after the onset of clinical manifestations, however, with only 32% of patients diagnosed on routine medical evaluation.[21]Adabag AS, Kuskowski MA, Maron BJ. Determinants for clinical diagnosis of hypertrophic cardiomyopathy. Am J Cardiol. 2006 Dec 1;98(11):1507-11.
http://www.ncbi.nlm.nih.gov/pubmed/17126660?tool=bestpractice.com
Laboratory DNA analysis for mutant genes is the most definitive method for establishing the diagnosis of hypertrophic cardiomyopathy (HCM), but is usually only used for screening purposes.
History
Family history
A family history of syncope, heart failure, or sudden or premature death should be taken. Sudden cardiac deaths may sometimes have been reported as accidental deaths, for example, drowning or unexplained traffic accidents.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Other points to note in the family history include cardiac transplantation, pacemaker and defibrillator implants, and features suggestive of systemic disease (e.g., stroke at a young age, skeletal muscle weakness, or renal disease). A three- to four-generation family pedigree should be created to aid in diagnosis, provide clues to etiology, determine inheritance pattern, and identify at-risk relatives.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Sarcomeric HCM is autosomal dominant and is therefore characterized by the presence of affected individuals across generations, with transmission from parents of either sex and a 50% risk of allele transmission to offspring. Family history may be negative, however, as the disease has incomplete penetrance.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Symptoms
Patients may be symptom-free. However, it is important to note any symptoms of presyncope or syncope, particularly when occurring with exercise, dyspnea on exertion, palpitations, or chest pain. Patients over 50 years of age may present with atrial fibrillation or symptoms of a stroke.[22]Robinson K, Frenneaux MP, Stockins B, et al. Atrial fibrillation in hypertrophic cardiomyopathy: a longitudinal study. J Am Coll Cardiol. 1990 May;15(6):1279-85.
https://www.jacc.org/doi/pdf/10.1016/S0735-1097%2810%2980014-2
http://www.ncbi.nlm.nih.gov/pubmed/2329232?tool=bestpractice.com
Physical examination
Examination may be remarkable for a left ventricular (LV) lift; a double apical impulse; a brisk carotid upstroke; a systolic ejection murmur at the lower left edge that is accentuated by exercise and standing and lessened by lying supine or squatting; and a fourth heart sound.[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
Diagnostic testing
ECG
A resting 12-lead ECG is recommended at the first clinic visit in all individuals with known or suspected HCM and should be repeated whenever there is a change in symptoms in patients with an established diagnosis.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Most patients have ECG abnormalities; these are not specific to HCM, but rather, should prompt further investigation with echocardiography. An abnormal ECG may predate the finding of hypertrophy on echocardiography.[23]Yetman AT, McCrindle BW. Management of pediatric hypertrophic cardiomyopathy. Curr Opin Cardiol. 2005 Mar;20(2):80-3.
http://www.ncbi.nlm.nih.gov/pubmed/15711191?tool=bestpractice.com
Repolarization abnormalities are common. T-wave inversion commonly involves the inferior and lateral leads and T waves are deep and often preceded by ST-segment depression.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
Deeply inverted T waves in the precordial leads are suggestive of apical HCM.[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
Prominent abnormal Q waves may be seen in the inferior (II, III, aVF) and/or lateral (I, aVL, V5-6) leads, reflecting septal hypertrophy.[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
Increased QRS voltages indicating left ventricular hypertrophy (LVH) may be present. These are nearly always associated with other ECG abnormalities in HCM.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
The presence of isolated QRS voltage criteria for LVH in the absence of other ECG markers is present in fewer than 2% of patients with HCM.[26]Bernardini A, Crotti L, Olivotto I, et al. Diagnostic and prognostic electrocardiographic features in patients with hypertrophic cardiomyopathy. Eur Heart J Suppl. 2023 May;25(suppl c):C173-8.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10132576
http://www.ncbi.nlm.nih.gov/pubmed/37125268?tool=bestpractice.com
ECG signs of left and right atrial enlargement and P-wave prolongation (a known predictor of atrial fibrillation) may be observed. They rarely occur in isolation; other ECG abnormalities such as repolarization changes or signs of LVH are generally present. Left atrial enlargement reflects diastolic dysfunction, high filling pressures, outflow obstruction, and functional mitral regurgitation. Left atrial dilatation and dysfunction are markers of adverse prognosis.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
Left-axis deviation (caused by LVH) and ventricular pre-excitation may also be seen.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
Some patients may present with arrhythmias, for example, atrial fibrillation or supraventricular tachycardia.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
The ECG is normal in only a small proportion (5% to 10%) of patients at presentation.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
These patients have been reported to have a more favorable clinical course than those with ECG abnormalities.[26]Bernardini A, Crotti L, Olivotto I, et al. Diagnostic and prognostic electrocardiographic features in patients with hypertrophic cardiomyopathy. Eur Heart J Suppl. 2023 May;25(suppl c):C173-8.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10132576
http://www.ncbi.nlm.nih.gov/pubmed/37125268?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: ECG showing changes associated with LVHFrom the collection of Melanie Everitt MD, Heart Failure & Transplantation Program, Primary Children's Medical Center, Salt Lake City, UT; used with permission [Citation ends]. [Figure caption and citation for the preceding image starts]: Giant T-wave inversionFrom the collection of Dr Anji T. Yetman MD, University of Utah; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Giant T-wave inversionFrom the collection of Dr Anji T. Yetman MD, University of Utah; used with permission [Citation ends].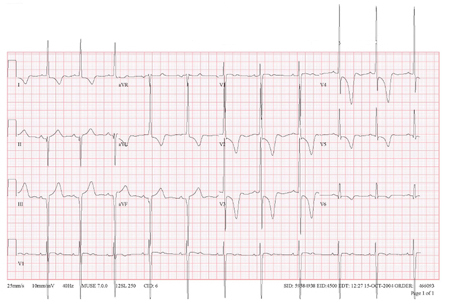
Laboratory tests
US guidelines do not recommend laboratory tests as part of the initial workup.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
However, European guidelines recommend that all patients with suspected or confirmed HCM should have routine laboratory tests done to establish etiology, assess disease severity, and aid in the detection of extracardiac manifestations and assessment of secondary organ dysfunction. The following first-line tests are recommended:[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Creatine kinase (CK): raised levels are a useful clue when trying to establish etiology; metabolic disorders such as Danon or mitochondrial disease, which can mimic HCM, should be considered. When CK is persistently raised, a detailed examination by a neurologist should be considered.
Liver function tests: liver dysfunction is prevalent in patients with chronic heart failure. Abnormal liver function tests can also be a useful clue when trying to establish etiology; metabolic disorders such as Danon disease, which can mimic HCM, should be considered.
Renal function: impaired renal function may be seen with severe LV dysfunction.
N-terminal pro-brain natriuretic peptide (NT-proBNP): high levels are associated with cardiovascular events, heart failure, and death, and may have diagnostic, prognostic, and therapeutic monitoring value.
Troponin: higher levels are associated with a higher risk of cardiovascular events, heart failure, and death, and may have diagnostic, prognostic, and therapeutic monitoring value.
Urinalysis; proteinuria is suggestive of renal impairment.
Following specialist evaluation, additional tests to detect rare metabolic and syndromic causes are often required in patients with cardiomyopathy and extracardiac features.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Chest x-ray
A chest x-ray may show cardiomegaly secondary to LVH or left atrial enlargement, or may be normal.[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
This test is not particularly sensitive.[Figure caption and citation for the preceding image starts]: CXR of a patient with HCM demonstrating cardiomegalyFrom the collection of Melanie Everitt MD, Heart Failure & Transplantation Program, Primary Children's Medical Center, Salt Lake City, UT; used with permission [Citation ends].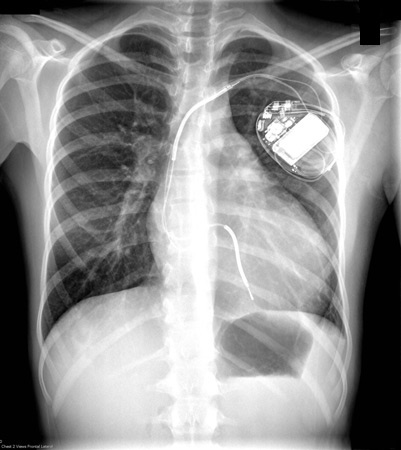
Transthoracic echocardiography
At initial assessment of all patients with HCM, transthoracic 2D and Doppler echocardiography are recommended. The classic finding is LVH, typically asymmetric hypertrophy of the septum.[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
Echocardiography is also used for family screening of an affected person, and for risk assessment of sudden cardiac death (SCD) in patients with a known diagnosis of HCM.
Clinical diagnosis of HCM is confirmed when maximal end-diastolic wall thickness ≥15 mm is imaged anywhere in the left ventricle (LV). More limited hypertrophy (≥13 mm) can be diagnostic when present in family members of a patient with HCM or in conjunction with a positive genetic test.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
Systolic anterior motion of the mitral valve may also be seen, along with mitral insufficiency.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
LV outflow tract obstruction (LVOTO) may be present. By convention, LVOTO is defined as a peak instantaneous Doppler LV outflow tract gradient of ≥30 mmHg, but the threshold for invasive treatment is usually considered to be ≥50 mmHg.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
There may be abnormalities of diastolic function (present in 80% of patients independent of the presence of LVOTO).[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
Diastolic dysfunction should be assessed by Doppler tissue imaging as part of the echocardiographic screening test of first-degree relatives, as this abnormality may precede the onset of overt LVH.[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
[28]Ho CY, Sweitzer NK, McDonough B, et al. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002 Jun 25;105(25):2992-7.
https://www.ahajournals.org/doi/10.1161/01.CIR.0000019070.70491.6D
http://www.ncbi.nlm.nih.gov/pubmed/12081993?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Apical 4-chamber image demonstrating hypertrophy of the interventricular septumFrom the collection of Dr Anji T. Yetman MD, University of Utah; used with permission [Citation ends]. [Figure caption and citation for the preceding image starts]: Long axis echocardiography view - asymmetric septal hypertrophyFrom the collection of Dr Anji T. Yetman MD, University of Utah; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Long axis echocardiography view - asymmetric septal hypertrophyFrom the collection of Dr Anji T. Yetman MD, University of Utah; used with permission [Citation ends].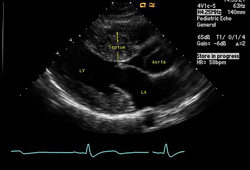
Other types of echocardiography
Stress echocardiography can be helpful in selected patients to evaluate myocardial ischemia.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Exercise echocardiography is useful to identify provocable LVOTO and exercise-induced mitral regurgitation in symptomatic patients with HCM.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Transesophageal echocardiography is limited to select indications, such as the exclusion of atrial thrombi related to atrial fibrillation, investigating the method of obstruction in patients with LVOTO where this is not obvious, elucidating the mechanism of mitral regurgitation, or planning invasive interventions such as septal myectomy.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Exercise ECG
Exercise testing is performed to aid in risk stratification. Abnormalities associated with an increased risk of SCD include abnormal blunted systolic BP response of <20 mmHg to exercise, ventricular arrhythmias, progressive ST depression, and symptoms.[29]Frenneaux MP. Assessing the risk of sudden cardiac death in a patient with hypertrophic cardiomyopathy. Heart. 2004 May;90(5):570-5.
https://heart.bmj.com/content/90/5/570
http://www.ncbi.nlm.nih.gov/pubmed/15084566?tool=bestpractice.com
[30]Yetman AT, McCrindle BW, MacDonald C, et al. Myocardial bridging in children with hypertrophic cardiomyopathy - a risk factor for sudden death. N Engl J Med. 1998 Oct 22;339(17):1201-9.
https://www.nejm.org/doi/full/10.1056/NEJM199810223391704
http://www.ncbi.nlm.nih.gov/pubmed/9780340?tool=bestpractice.com
Holter monitoring
This may be normal or demonstrate ventricular or supraventricular arrhythmias. Ventricular arrhythmias are associated with an increased risk of sudden death.[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
Nuclear medicine techniques
Patients with exertional chest pain or ventricular tachycardia on Holter monitoring should undergo nuclear testing with either single-photon-emission computed tomography or positron emission tomography.[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
Myocardial perfusion imaging may demonstrate perfusion defects even in the absence of obstructive lesions.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
Patients may have fixed or reversible defects. Patients with reversible defects should undergo cardiac catheterization to identify possible causes of ischemia.
Nuclear medicine can also play a role in diagnosis; it is particularly helpful in the etiological diagnosis of cardiac amyloidosis.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Cardiac magnetic resonance (CMR)
Contrast-enhanced CMR can be a useful adjunct in patients with HCM at initial evaluation.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
It can aid diagnosis and contribute to risk stratification and management.
LV wall thickness can be assessed; the use of CMR may thus increase the diagnostic yield in patients with suspected HCM who have poor visualization by echocardiogram of the LV walls or apex.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
Systolic and diastolic function can also be assessed, as well as mitral valve function, LVOTO, and left atrial dimensions.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
The use of late gadolinium enhancement techniques can identify areas of myocardial fibrosis that may be a marker for adverse outcomes, or may aid in differentiating HCM from an athletic heart.[31]O’Hanlon R, Assomull RG, Prasad SK. Use of cardiovascular magnetic resonance for diagnosis and management in hypertrophic cardiomyopathy. Curr Cardiol Rep. 2007 Mar;9(1):51-6.
http://www.ncbi.nlm.nih.gov/pubmed/17362685?tool=bestpractice.com
CMR is also emerging as a means of identifying patients who are at increased risk for arrhythmias. Several studies have found the presence of myocardial fibrosis by late gadolinium enhancement to be associated with the occurrence of ventricular arrhythmias, as well as an independent risk factor for death.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[32]Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):875-87.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.007
http://www.ncbi.nlm.nih.gov/pubmed/20667520?tool=bestpractice.com
[33]Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008 Apr 8;51(14):1369-74.
http://www.ncbi.nlm.nih.gov/pubmed/18387438?tool=bestpractice.com
[34]Fluechter S, Kuschyk J, Wolpert C, et al. Extent of late gadolinium enhancement detected by cardiovascular magnetic resonance correlates with the inducibility of ventricular tachyarrhythmia in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2010 May 21;12:30.
https://jcmr-online.biomedcentral.com/articles/10.1186/1532-429X-12-30
http://www.ncbi.nlm.nih.gov/pubmed/20492668?tool=bestpractice.com
[35]Suk T, Edwards C, Hart H, et al. Myocardial scar detected by contrast-enhanced cardiac magnetic resonance imaging is associated with ventricular tachycardia in hypertrophic cardiomyopathy patients. Heart Lung Circ. 2008 Oct;17(5):370-4.
http://www.ncbi.nlm.nih.gov/pubmed/18562248?tool=bestpractice.com
[36]Leonardi S, Raineri C, De Ferrari GM, et al. Usefulness of cardiac magnetic resonance in assessing the risk of ventricular arrhythmias and sudden death in patients with hypertrophic cardiomyopathy. Eur Heart J. 2009 Aug;30(16):2003-10.
https://academic.oup.com/eurheartj/article/30/16/2003/630669
http://www.ncbi.nlm.nih.gov/pubmed/19474054?tool=bestpractice.com
[37]O'Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):867-74.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.010
http://www.ncbi.nlm.nih.gov/pubmed/20688032?tool=bestpractice.com
Tissue characterization on CMR can provide clues regarding etiology, as characteristic findings are associated with certain diseases, for example, in sarcomeric HCM, a patchy mid-wall in hypertrophied areas is typical, while in amyloidosis-related cardiac hypertrophy, diffuse subendocardial late gadolinium enhancement is seen. These findings should be assessed collectively with genetic results and other clinical features by operators expert in cardiac imaging and the evaluation of heart muscle disease.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Serial follow-up CMR, every 2-5 years depending on initial severity and clinical course, can assist in evaluating disease progression as well as the benefits of therapy.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Cardiac computed tomography (CT)
Although not used commonly, CT can provide important insights when echocardiography is technically limited and CMR imaging is contraindicated or unavailable.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
Cardiac CT provides clear definition of LV structure (including hypertrophy pattern, wall thickness measurement, detection of subaortic membrane, and intracardiac thrombus) and function. Disadvantages of CT are the use of radiation and radioiodine contrast and inferior temporal resolution compared with echocardiography.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
CT coronary angiography
Indicated in patients with exertional chest pain or ischemia on nuclear testing to check for the presence of concomitant coronary artery disease or myocardial bridging, comorbidities which can affect the clinical manifestations and course of HCM.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
Cardiac catheterization
In symptomatic patients with HCM and inconclusive noninvasive cardiac imaging, left and right heart catheterization may be considered to assess the severity of LVOTO and to measure LV filling pressures.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
It can also be used to check for the presence of concomitant coronary disease or myocardial bridging, and is recommended for patients who are candidates for septal reduction therapy.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
Endomyocardial biopsy
Endomyocardial biopsy is not usually recommended for diagnosis of HCM but may be considered on rare occasions, especially when the pattern of hypertrophy is diffuse and there is suspicion for other cardiomyopathies presenting with hypertrophy.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Risk stratification
Risk of sudden cardiac death (SCD)
After diagnosis, patients should undergo risk stratification including Holter monitoring and exercise ECG, unless contraindicated, to further define their risk of sudden death.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
The European Society of Cardiology (ESC) has developed a risk prediction calculator for SCD at 5 years in patients ≥16 years with HCM.[38]O'Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014 Aug 7;35(30):2010-20.
https://academic.oup.com/eurheartj/article/35/30/2010/467191
http://www.ncbi.nlm.nih.gov/pubmed/24126876?tool=bestpractice.com
Risk calculation is based on age, maximal LV wall thickness, left atrial diameter, LV outflow tract gradient, family history of SCD, nonsustained ventricular tachycardia, and unexplained syncope. Risk of SCD should be reevaluated at 1-2 year intervals or whenever there is a change in clinical status.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
These models should not be used in elite athletes or in individuals with metabolic/infiltrative diseases (e.g., Anderson-Fabry disease) and syndromes (e.g., Noonan syndrome).[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
It should be noted that current risk-stratification models may be unreliable in the prediction of future sudden death, and implantable cardioverter-defibrillator (ICD) placement may still be warranted in HCM patients with low-risk scores.[39]Maron BJ, Casey SA, Chan RH, et al. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol. 2015 Sep 1;116(5):757-64.
http://www.ncbi.nlm.nih.gov/pubmed/26183790?tool=bestpractice.com
US guidelines reflect this uncertainty, recommending that the tool be used as an aid to the shared decision-making process for ICD placement in patients with clinical risk markers, whereas European guidelines recommend that it should be used as the basis of decision-making for all patients with HCM.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
ESC guidelines recommend that implantation of an ICD should be considered in patients with an estimated 5-year risk of sudden death of ≥6%, following detailed clinical assessment that considers: (i) the lifelong risk of complications; (ii) competing mortality risk from the disease and comorbidities; AND (iii) the impact of an ICD on lifestyle, socioeconomic status, and psychological health. It may also be considered in patients with a risk between ≥4% and <6% on an individual basis. For patients who are in the low-risk category (<4% estimated 5-year risk of SCD), ICD is generally not recommended. However, the guidelines acknowledge that ICD may be considered in low-risk patients who have extensive late gadolinium enhancement (≥15%) on CMR or LV ejection fraction <50%; these factors do not form part of the HCM risk-SCD, but evidence suggests that they increase the risk of SCD. Shared decision-making is recommended.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Risk factors for SCD are as follows:[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[40]Goldberger JJ, Cain ME, Hohnloser SH, et al. American Heart Association/American College of Cardiology Foundation/Heart Rhythm Society scientific statement on noninvasive risk stratification techniques for identifying patients at risk for sudden cardiac death. J Am Coll Cardiol. 2008 Sep 30;52(14):1179-99.
https://www.jacc.org/doi/10.1016/j.jacc.2008.05.003
http://www.ncbi.nlm.nih.gov/pubmed/18804749?tool=bestpractice.com
Younger age: some studies have reported a significantly increased risk of SCD in younger patients.
Nonsustained ventricular tachycardia (defined as ≥3 consecutive ventricular beats at ≥120 beats per minute lasting <30 seconds) on Holter monitor: occurs in 20% to 30% of patients.
Abnormal blood pressure (BP) response to exercise: defined as a rise in systolic BP of <20 mmHg, no rise, or a fall in BP of >20 mmHg during exercise. Medications may affect the BP response and should be considered in the interpretation of exercise test results.
Massive hypertrophy (LV wall thickness ≥30 mm).
Severe LVOTO by echocardiogram (LVOTO >30 to 50 mmHg): while severe obstruction is considered a minor risk factor for sudden death, the degree of outflow tract obstruction generally does not correlate with the risk of sudden death. Medical therapy or surgery to decrease outflow tract obstruction does not decrease the risk of sudden death.
Family history of sudden death: while definitions vary, a family history of SCD is usually considered clinically significant when one or more first-degree relatives have died suddenly ages <40 years with or without a diagnosis of HCM, or when SCD has occurred in a first-degree relative at any age with an established diagnosis of HCM.
Personal history of unexplained syncope.
Prior cardiac arrest, or sustained ventricular tachycardia.
LV systolic dysfunction with ejection fraction <50%.
Left atrial enlargement.
Presence of LV apical aneurysm.
Diffuse and extensive late gadolinium enhancement by cardiac MRI: cardiac magnetic resonance imaging is emerging as a means of identifying patients who are at increased risk for arrhythmias. Several studies have found the presence of myocardial fibrosis by late gadolinium enhancement to be associated with the occurrence of ventricular arrhythmias.[33]Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008 Apr 8;51(14):1369-74.
http://www.ncbi.nlm.nih.gov/pubmed/18387438?tool=bestpractice.com
[34]Fluechter S, Kuschyk J, Wolpert C, et al. Extent of late gadolinium enhancement detected by cardiovascular magnetic resonance correlates with the inducibility of ventricular tachyarrhythmia in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2010 May 21;12:30.
https://jcmr-online.biomedcentral.com/articles/10.1186/1532-429X-12-30
http://www.ncbi.nlm.nih.gov/pubmed/20492668?tool=bestpractice.com
[35]Suk T, Edwards C, Hart H, et al. Myocardial scar detected by contrast-enhanced cardiac magnetic resonance imaging is associated with ventricular tachycardia in hypertrophic cardiomyopathy patients. Heart Lung Circ. 2008 Oct;17(5):370-4.
http://www.ncbi.nlm.nih.gov/pubmed/18562248?tool=bestpractice.com
[36]Leonardi S, Raineri C, De Ferrari GM, et al. Usefulness of cardiac magnetic resonance in assessing the risk of ventricular arrhythmias and sudden death in patients with hypertrophic cardiomyopathy. Eur Heart J. 2009 Aug;30(16):2003-10.
https://academic.oup.com/eurheartj/article/30/16/2003/630669
http://www.ncbi.nlm.nih.gov/pubmed/19474054?tool=bestpractice.com
[37]O'Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):867-74.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.010
http://www.ncbi.nlm.nih.gov/pubmed/20688032?tool=bestpractice.com
The presence of fibrosis has also been found to be an independent risk for death.[32]Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):875-87.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.007
http://www.ncbi.nlm.nih.gov/pubmed/20667520?tool=bestpractice.com
Investigation for ischemia
Ischemia may be related to myocardial bridging, LVOTO, or massive hypertrophy with reduced myocardial perfusion. The presence of ischemia is a weak risk factor for SCD. Patients with angina or ST depression on exercise ECG should be evaluated for ischemia with nuclear imaging or CT arteriography if the likelihood of coronary artery disease (CAD) is relatively low. CT arteriography or cardiac catheterization are indicated if there is a higher likelihood of CAD given other patient factors.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[41]Lubarsky L, Gupta MP, Hecht HS. Evaluation of myocardial bridging of the left anterior descending coronary artery by 64-slice multidetector computed tomographic angiography. Am J Cardiol. 2007 Oct 1;100(7):1081-2.
http://www.ncbi.nlm.nih.gov/pubmed/17884365?tool=bestpractice.com
Myocardial bridging (tunneling of coronary arteries into the heart muscle) should also be considered in the setting of angina or ischemia. Cardiac catheterization or CT arteriography can be used to evaluate bridging with specific attention given by the interpreter to this possible diagnosis.[42]Kantarci M, Doganay S, Karcaaltincaba M, et al. Clinical situations in which coronary CT angiography confers superior diagnostic information compared with coronary angiography. Diagn Interv Radiol. 2012 May-Jun;18(3):261-9.
https://www.dirjournal.org/en/clinical-situations-in-which-coronary-ct-angiography-confers-superior-diagnostic-information-compared-with-coronary-angiography-166357
http://www.ncbi.nlm.nih.gov/pubmed/22261852?tool=bestpractice.com
Genetic mutation analysis
Genetic testing in an individual with cardiomyopathy (known as confirmatory testing or diagnostic testing) is recommended for their direct benefit: (i) to confirm the diagnosis; (ii) where it may inform prognosis; (iii) where it may inform treatment selection; or (iv) where it may inform their reproductive management. Genetic testing of an affected individual may also be indicated if there are relatives who may benefit from testing, particularly those who will be enrolled in long-term surveillance if the genetic etiology is not established (and who may be spared this burden if a genetic diagnosis is made in the family).[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
The clinical utility of genetic testing has limitations. Currently identified disease-causing genes are thought to account for only 80% of cases. Moreover, the sensitivity of commercially available genetic testing depends on the number of genes screened for by the particular laboratory and may be <80%. When the 8 most common sarcomeric mutations are screened, the clinical sensitivity approaches 60%.[17]Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008 Jan;19(1):104-10.
https://onlinelibrary.wiley.com/doi/10.1111/j.1540-8167.2007.00965.x
http://www.ncbi.nlm.nih.gov/pubmed/17916152?tool=bestpractice.com
In up to 40% of patients with HCM, no sarcomere variant is identified, and there is no family history of disease.[43]Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022 Sep 1;24(8):1307-67.
https://academic.oup.com/europace/article/24/8/1307/6562982?login=false
http://www.ncbi.nlm.nih.gov/pubmed/35373836?tool=bestpractice.com
The absence of a monogenic disease-causing variant on conventional genetic testing leaves three possibilities: (i) either there is a monogenic cause that has not been identified (i.e., not detected or recognized as causative by current testing); (ii) the cardiomyopathy does not have a genetic etiology; or (iii) the cardiomyopathy is attributable to the effects of multiple variants of individually smaller effect.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Despite its limitations, genetic testing is of value in screening family members of an affected patient with an identified mutation. Genetic testing in this situation will determine who requires ongoing clinical evaluation (known as cascade testing):[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[43]Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022 Sep 1;24(8):1307-67.
https://academic.oup.com/europace/article/24/8/1307/6562982?login=false
http://www.ncbi.nlm.nih.gov/pubmed/35373836?tool=bestpractice.com
[44]Musunuru K, Hershberger RE, Day SM, et al. Genetic testing for inherited cardiovascular diseases: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2020 Aug;13(4):e000067.
https://www.ahajournals.org/doi/10.1161/HCG.0000000000000067
http://www.ncbi.nlm.nih.gov/pubmed/32698598?tool=bestpractice.com
Relatives with the identified mutation should continue to be screened for the clinical development of HCM. The development of clinically apparent disease may occur late in adulthood, so screening should be lifelong.
Gene-negative relatives can be reassured that they do not have the disease-causing mutation and do not require further screening.
While identification of a gene mutation during cascade testing indicates that the development of HCM is very likely, genotype-phenotype variability exists. Despite identical gene mutations, the gene mutation may manifest as HCM, restrictive cardiomyopathy, dilated cardiomyopathy, or no clinically apparent abnormality in different patients. For those without apparent disease, late-onset penetrance must be considered.[45]Christiaans I, Birnie E, van Langen IM, et al. The yield of risk stratification for sudden cardiac death in hypertrophic cardiomyopathy myosin-binding protein C gene mutation carriers: focus on predictive screening. Eur Heart J. 2010 Apr;31(7):842-8.
https://academic.oup.com/eurheartj/article/31/7/842/431846
http://www.ncbi.nlm.nih.gov/pubmed/20019025?tool=bestpractice.com
The risk of sudden death may be low or high for the same mutation.[46]Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010 Dec 7;122(23):2430-40.
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.110.978924
http://www.ncbi.nlm.nih.gov/pubmed/21135371?tool=bestpractice.com
Genetic counseling should be available for all patients who are offered genetic testing, to inform decision-making and ensure so that results can be reviewed and their clinical significance appropriately determined.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
The importance of potential psychologic, social, legal, ethical, and professional implications of having a genetic disease should also be discussed, and appropriate support provided.[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
Prenatal genetic counseling should be offered to parents who have had a previous affected child with an inherited HCM due to a single or multiple pathogenic variant(s), or to couples where one or both partners carries a known pathogenic variant.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[47]Joglar JA, Kapa S, Saarel EV, et al. 2023 HRS expert consensus statement on the management of arrhythmias during pregnancy. Heart Rhythm. 2023 Oct;20(10):e175-e264.
https://www.heartrhythmjournal.com/article/S1547-5271(23)02246-4/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/37211147?tool=bestpractice.com
The risk of disease transmission should be discussed, as well as potential reproductive options (e.g., in-vitro fertilization with preimplantation genetic diagnosis, prenatal genetic screening, and postpartum genetic testing).[2]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020 Dec 22;142(25):e558-631.
https://www.ahajournals.org/doi/10.1161/CIR.0000000000000937
http://www.ncbi.nlm.nih.gov/pubmed/33215931?tool=bestpractice.com
[43]Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022 Sep 1;24(8):1307-67.
https://academic.oup.com/europace/article/24/8/1307/6562982?login=false
http://www.ncbi.nlm.nih.gov/pubmed/35373836?tool=bestpractice.com
Advances in genetic sequencing technology and increased accessibility to testing have led to an increasing number of incidentally identified genetic variants associated with HCM. Interpreting the clinical relevance of such findings can be challenging; the American Heart Association has produced guidance on how to manage them, with emphasis on a multidisciplinary team approach.[48]Landstrom AP, Chahal AA, Ackerman MJ, et al. Interpreting incidentally identified variants in genes associated with heritable cardiovascular disease: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2023 Apr;16(2):e000092.
https://www.ahajournals.org/doi/full/10.1161/HCG.0000000000000092?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/36970980?tool=bestpractice.com
The American College of Medical Genetics and Genomics has recommended that cardiomyopathy-associated genes be evaluated for secondary findings whenever broad clinical sequencing is undertaken, regardless of the initial indication for testing.[49]National Library of Medicine. ACMG recommendations for reporting of secondary findings in clinical exome and genome sequencing [internet publication].
https://www.ncbi.nlm.nih.gov/clinvar/docs/acmg
There is currently no international consensus around this recommendation, however.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
