Introduction
Diagnosis of adult-onset Still disease (AOSD) is often difficult because it has a variable course and the clinical features overlap with those seen in a wide range of other inflammatory conditions.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
As there is no definitive diagnostic test for AOSD, it is often a diagnosis of exclusion.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[22]National Health Service. Clinical Commissioning Policy: tocilizumab for the treatment of adult-onset Still’s disease refractory to second-line therapy (adults) [210801P] (URN: 1609). 2021 [internet publication].
https://www.england.nhs.uk/wp-content/uploads/2021/08/1609-Tocilizumab-for-AOSD-Final-August-2021-.pdf
Due to this difficulty, diagnosis is frequently delayed.[23]Franchini S, Dagna L, Salvo F, et al. Adult onset Still's disease: clinical presentation in a large cohort of Italian patients. Clin Exp Rheumatol. 2010 Jan-Feb;28(1):41-8.
https://www.clinexprheumatol.org/abstract.asp?a=433
http://www.ncbi.nlm.nih.gov/pubmed/20346237?tool=bestpractice.com
[24]Gerfaud-Valentin M, Jamilloux Y, Iwaz J, et al. Adult-onset Still's disease. Autoimmun Rev. 2014 Jul;13(7):708-22.
https://www.doi.org/10.1016/j.autrev.2014.01.058
http://www.ncbi.nlm.nih.gov/pubmed/24657513?tool=bestpractice.com
[25]Hu QY, Zeng T, Sun CY, et al. Clinical features and current treatments of adult-onset Still's disease: a multicentre survey of 517 patients in China. Clin Exp Rheumatol. 2019 Nov-Dec;37(6 suppl 121):52-7.
https://www.clinexprheumatol.org/abstract.asp?a=13551
http://www.ncbi.nlm.nih.gov/pubmed/31573475?tool=bestpractice.com
[26]Kim YJ, Koo BS, Kim YG, et al. Clinical features and prognosis in 82 patients with adult-onset Still's disease. Clin Exp Rheumatol. 2014 Jan-Feb;32(1):28-33.
http://www.ncbi.nlm.nih.gov/pubmed/24050706?tool=bestpractice.com
[27]Pay S, Türkçapar N, Kalyoncu M, et al. A multicenter study of patients with adult-onset Still's disease compared with systemic juvenile idiopathic arthritis. Clin Rheumatol. 2006 Sep;25(5):639-44.
https://www.doi.org/10.1007/s10067-005-0138-5
http://www.ncbi.nlm.nih.gov/pubmed/16365690?tool=bestpractice.com
Consider the diagnosis of AOSD if a young adult presents with daily intermittent high fevers, arthralgia, and a salmon-pink skin rash.
Presentation varies and may include additional systemic features such as pharyngitis, pleuritis, or pericarditis. It may be a first episode, or one of intermittent or prolonged chronic symptoms.
Check CBC, CRP, ferritin, and, if available, glycosylated ferritin. These are key investigations alongside others to rule out differentials such as infection, malignancy, or autoimmune disease.
Consider the clinical findings and investigation results alongside diagnostic classification criteria to help identify the condition.
History
Take a thorough history. Ask about the following most common features:[1]Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018 Sep;93:24-36.
https://www.doi.org/10.1016/j.jaut.2018.07.018
http://www.ncbi.nlm.nih.gov/pubmed/30077425?tool=bestpractice.com
[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Fever
Almost all patients experience fever, which is typically high spiking (≥102.2°F [≥39.0°C]), occurring daily or occasionally twice daily over a period of at least 1 week, and resolving each time within a few hours.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Larger, more recent studies report fever in 91% to 100% of people with AOSD.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[7]Sfriso P, Priori R, Valesini G, et al. Adult-onset Still's disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016 Jul;35(7):1683-9.
https://www.doi.org/10.1007/s10067-016-3308-8
http://www.ncbi.nlm.nih.gov/pubmed/27207567?tool=bestpractice.com
[25]Hu QY, Zeng T, Sun CY, et al. Clinical features and current treatments of adult-onset Still's disease: a multicentre survey of 517 patients in China. Clin Exp Rheumatol. 2019 Nov-Dec;37(6 suppl 121):52-7.
https://www.clinexprheumatol.org/abstract.asp?a=13551
http://www.ncbi.nlm.nih.gov/pubmed/31573475?tool=bestpractice.com
[28]Zhang Y, Yang Y, Bai Y, et al. Clinical characteristics and follow-up analysis of adult-onset Still's disease complicated by hemophagocytic lymphohistiocytosis. Clin Rheumatol. 2016 May;35(5):1145-51.
https://www.doi.org/10.1007/s10067-016-3178-0
http://www.ncbi.nlm.nih.gov/pubmed/26809798?tool=bestpractice.com
[29]Asanuma YF, Mimura T, Tsuboi H, et al. Nationwide epidemiological survey of 169 patients with adult Still's disease in Japan. Mod Rheumatol. 2015 May;25(3):393-400.
https://www.doi.org/10.3109/14397595.2014.974881
http://www.ncbi.nlm.nih.gov/pubmed/25382730?tool=bestpractice.com
[30]Kalyoncu U, Solmaz D, Emmungil H, et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still's disease: data from a large multicenter cohort. J Autoimmun. 2016 May;69:59-63.
https://www.doi.org/10.1016/j.jaut.2016.02.010
http://www.ncbi.nlm.nih.gov/pubmed/26970681?tool=bestpractice.com
[31]Nakamura H, Fujieda Y, Tarumi M, et al. Calcineurin inhibitors for adult-onset Still's disease: a multicentre retrospective cohort study. Clin Exp Rheumatol. 2020 Sep-Oct;38(5 suppl 127):11-6.
http://www.ncbi.nlm.nih.gov/pubmed/32083551?tool=bestpractice.com
Arthralgia and arthritis
Arthralgia is reported in 47% to 95% in different studies of people with AOSD.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[7]Sfriso P, Priori R, Valesini G, et al. Adult-onset Still's disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016 Jul;35(7):1683-9.
https://www.doi.org/10.1007/s10067-016-3308-8
http://www.ncbi.nlm.nih.gov/pubmed/27207567?tool=bestpractice.com
[25]Hu QY, Zeng T, Sun CY, et al. Clinical features and current treatments of adult-onset Still's disease: a multicentre survey of 517 patients in China. Clin Exp Rheumatol. 2019 Nov-Dec;37(6 suppl 121):52-7.
https://www.clinexprheumatol.org/abstract.asp?a=13551
http://www.ncbi.nlm.nih.gov/pubmed/31573475?tool=bestpractice.com
[28]Zhang Y, Yang Y, Bai Y, et al. Clinical characteristics and follow-up analysis of adult-onset Still's disease complicated by hemophagocytic lymphohistiocytosis. Clin Rheumatol. 2016 May;35(5):1145-51.
https://www.doi.org/10.1007/s10067-016-3178-0
http://www.ncbi.nlm.nih.gov/pubmed/26809798?tool=bestpractice.com
[29]Asanuma YF, Mimura T, Tsuboi H, et al. Nationwide epidemiological survey of 169 patients with adult Still's disease in Japan. Mod Rheumatol. 2015 May;25(3):393-400.
https://www.doi.org/10.3109/14397595.2014.974881
http://www.ncbi.nlm.nih.gov/pubmed/25382730?tool=bestpractice.com
[30]Kalyoncu U, Solmaz D, Emmungil H, et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still's disease: data from a large multicenter cohort. J Autoimmun. 2016 May;69:59-63.
https://www.doi.org/10.1016/j.jaut.2016.02.010
http://www.ncbi.nlm.nih.gov/pubmed/26970681?tool=bestpractice.com
[31]Nakamura H, Fujieda Y, Tarumi M, et al. Calcineurin inhibitors for adult-onset Still's disease: a multicentre retrospective cohort study. Clin Exp Rheumatol. 2020 Sep-Oct;38(5 suppl 127):11-6.
http://www.ncbi.nlm.nih.gov/pubmed/32083551?tool=bestpractice.com
Arthritis (seen in 51% to 66% of patients) typically begins as mild and localized, but it can become increasingly severe and polyarticular over the course of the disease.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Joints that are commonly affected include the proximal interphalangeal joints, wrists, elbows, knees, and ankles.[23]Franchini S, Dagna L, Salvo F, et al. Adult onset Still's disease: clinical presentation in a large cohort of Italian patients. Clin Exp Rheumatol. 2010 Jan-Feb;28(1):41-8.
https://www.clinexprheumatol.org/abstract.asp?a=433
http://www.ncbi.nlm.nih.gov/pubmed/20346237?tool=bestpractice.com
[25]Hu QY, Zeng T, Sun CY, et al. Clinical features and current treatments of adult-onset Still's disease: a multicentre survey of 517 patients in China. Clin Exp Rheumatol. 2019 Nov-Dec;37(6 suppl 121):52-7.
https://www.clinexprheumatol.org/abstract.asp?a=13551
http://www.ncbi.nlm.nih.gov/pubmed/31573475?tool=bestpractice.com
[32]Liu Z, Lv X, Tang G. Clinical features and prognosis of adult-onset Still's disease: 75 cases from China. Int J Clin Exp Med. 2015;8(9):16634-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4659083
http://www.ncbi.nlm.nih.gov/pubmed/26629195?tool=bestpractice.com
[33]Riera E, Olivé A, Narváez J, et al. Adult onset Still's disease: review of 41 cases. Clin Exp Rheumatol. 2011 Mar-Apr;29(2):331-6.
http://www.ncbi.nlm.nih.gov/pubmed/21385548?tool=bestpractice.com
[34]Zeng T, Zou YQ, Wu MF, et al. Clinical features and prognosis of adult-onset still's disease: 61 cases from China. J Rheumatol. 2009 May;36(5):1026-31.
https://www.doi.org/10.3899/jrheum.080365
http://www.ncbi.nlm.nih.gov/pubmed/19273456?tool=bestpractice.com
[35]Chen DY, Lan JL, Hsieh TY, et al. Clinical manifestations, disease course, and complications of adult-onset Still's disease in Taiwan. J Formos Med Assoc. 2004 Nov;103(11):844-52.
http://www.ncbi.nlm.nih.gov/pubmed/15549152?tool=bestpractice.com
The distal interphalangeal joints and shoulder joints are typically spared.[23]Franchini S, Dagna L, Salvo F, et al. Adult onset Still's disease: clinical presentation in a large cohort of Italian patients. Clin Exp Rheumatol. 2010 Jan-Feb;28(1):41-8.
https://www.clinexprheumatol.org/abstract.asp?a=433
http://www.ncbi.nlm.nih.gov/pubmed/20346237?tool=bestpractice.com
[25]Hu QY, Zeng T, Sun CY, et al. Clinical features and current treatments of adult-onset Still's disease: a multicentre survey of 517 patients in China. Clin Exp Rheumatol. 2019 Nov-Dec;37(6 suppl 121):52-7.
https://www.clinexprheumatol.org/abstract.asp?a=13551
http://www.ncbi.nlm.nih.gov/pubmed/31573475?tool=bestpractice.com
[32]Liu Z, Lv X, Tang G. Clinical features and prognosis of adult-onset Still's disease: 75 cases from China. Int J Clin Exp Med. 2015;8(9):16634-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4659083
http://www.ncbi.nlm.nih.gov/pubmed/26629195?tool=bestpractice.com
[33]Riera E, Olivé A, Narváez J, et al. Adult onset Still's disease: review of 41 cases. Clin Exp Rheumatol. 2011 Mar-Apr;29(2):331-6.
http://www.ncbi.nlm.nih.gov/pubmed/21385548?tool=bestpractice.com
[34]Zeng T, Zou YQ, Wu MF, et al. Clinical features and prognosis of adult-onset still's disease: 61 cases from China. J Rheumatol. 2009 May;36(5):1026-31.
https://www.doi.org/10.3899/jrheum.080365
http://www.ncbi.nlm.nih.gov/pubmed/19273456?tool=bestpractice.com
[35]Chen DY, Lan JL, Hsieh TY, et al. Clinical manifestations, disease course, and complications of adult-onset Still's disease in Taiwan. J Formos Med Assoc. 2004 Nov;103(11):844-52.
http://www.ncbi.nlm.nih.gov/pubmed/15549152?tool=bestpractice.com
Polyarticular disease is more common (30% to 90%) than oligoarticular (2% to 42%) or monoarticular (2% to 12%).[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
Skin rash
The typical skin rash in AOSD is salmon-pink, nonpruritic, and maculopapular. It is transient, occurring during fever. Larger, more recent studies report a frequency of 62% to 80%.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[7]Sfriso P, Priori R, Valesini G, et al. Adult-onset Still's disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016 Jul;35(7):1683-9.
https://www.doi.org/10.1007/s10067-016-3308-8
http://www.ncbi.nlm.nih.gov/pubmed/27207567?tool=bestpractice.com
[25]Hu QY, Zeng T, Sun CY, et al. Clinical features and current treatments of adult-onset Still's disease: a multicentre survey of 517 patients in China. Clin Exp Rheumatol. 2019 Nov-Dec;37(6 suppl 121):52-7.
https://www.clinexprheumatol.org/abstract.asp?a=13551
http://www.ncbi.nlm.nih.gov/pubmed/31573475?tool=bestpractice.com
[28]Zhang Y, Yang Y, Bai Y, et al. Clinical characteristics and follow-up analysis of adult-onset Still's disease complicated by hemophagocytic lymphohistiocytosis. Clin Rheumatol. 2016 May;35(5):1145-51.
https://www.doi.org/10.1007/s10067-016-3178-0
http://www.ncbi.nlm.nih.gov/pubmed/26809798?tool=bestpractice.com
[29]Asanuma YF, Mimura T, Tsuboi H, et al. Nationwide epidemiological survey of 169 patients with adult Still's disease in Japan. Mod Rheumatol. 2015 May;25(3):393-400.
https://www.doi.org/10.3109/14397595.2014.974881
http://www.ncbi.nlm.nih.gov/pubmed/25382730?tool=bestpractice.com
[30]Kalyoncu U, Solmaz D, Emmungil H, et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still's disease: data from a large multicenter cohort. J Autoimmun. 2016 May;69:59-63.
https://www.doi.org/10.1016/j.jaut.2016.02.010
http://www.ncbi.nlm.nih.gov/pubmed/26970681?tool=bestpractice.com
[31]Nakamura H, Fujieda Y, Tarumi M, et al. Calcineurin inhibitors for adult-onset Still's disease: a multicentre retrospective cohort study. Clin Exp Rheumatol. 2020 Sep-Oct;38(5 suppl 127):11-6.
http://www.ncbi.nlm.nih.gov/pubmed/32083551?tool=bestpractice.com
Other reported skin manifestations include urticaria and dermatographism (seen in 31% to 59% of patients).[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
These are pruritic hives that appear and resolve over minutes to hours, leaving normal skin behind. In the case of dermatographism, the hives typically arise from scratching or mild trauma of the skin. Neutrophilic urticarial dermatosis may also occur, consisting of urticarial lesions lasting 24-48 hours with dense neutrophilic infiltrates on biopsy.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Salmon-pink rash typical of AOSD on right arm of a 28-year-old woman of Nigerian heritageAkintayo RO et al. BMJ Case Reports 2015; 2015: bcr2015210789; used with permission [Citation ends].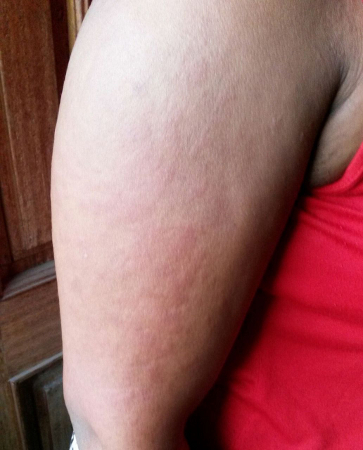 [Figure caption and citation for the preceding image starts]: Salmon-pink rash on the chest and neck of a white man with AOSDFrom the collection of Dr Sinisa Savic; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Salmon-pink rash on the chest and neck of a white man with AOSDFrom the collection of Dr Sinisa Savic; used with permission [Citation ends].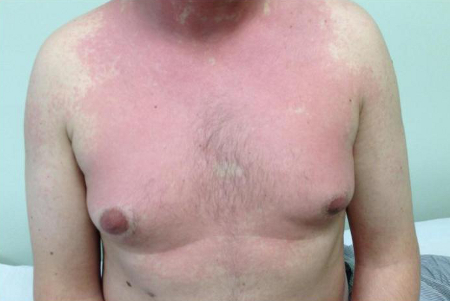
Other common symptoms
Ask about other common symptoms of AOSD including the following, which typically occur during fevers:
Sore throat/pharyngitis: seen in over half of patients and up to 92% in some studies, but less common in patients ages ≥65 years (54% compared with 86% in one study).[32]Liu Z, Lv X, Tang G. Clinical features and prognosis of adult-onset Still's disease: 75 cases from China. Int J Clin Exp Med. 2015;8(9):16634-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4659083
http://www.ncbi.nlm.nih.gov/pubmed/26629195?tool=bestpractice.com
[36]Kong XD, Xu D, Zhang W, et al. Clinical features and prognosis in adult-onset Still's disease: a study of 104 cases. Clin Rheumatol. 2010 Sep;29(9):1015-9.
http://www.ncbi.nlm.nih.gov/pubmed/20549276?tool=bestpractice.com
[37]Kishida D, Ichikawa T, Takamatsu R, et al. Clinical characteristics and treatment of elderly onset adult-onset Still's disease. Sci Rep. 2022 Apr 26;12(1):6787.
https://www.doi.org/10.1038/s41598-022-10932-3
http://www.ncbi.nlm.nih.gov/pubmed/35474094?tool=bestpractice.com
Myalgia: frequency: 26% to 53%[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Pleuritis: different studies report variable incidence, with some reporting it in over half of patients with AOSD.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
In practice, patients sometimes have a history of unverified reports of high temperatures, repeated courses of antibiotics for sore throats, and a rash that can settle with the fever, leaving little to see for an attending physician.
Other less common features include symptoms associated with pericarditis and myocarditis.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
Older patients ages ≥65 years present similarly, although they are less likely than younger patients to develop pharyngitis and more likely to develop pleuritis (46% vs. 17%).[37]Kishida D, Ichikawa T, Takamatsu R, et al. Clinical characteristics and treatment of elderly onset adult-onset Still's disease. Sci Rep. 2022 Apr 26;12(1):6787.
https://www.doi.org/10.1038/s41598-022-10932-3
http://www.ncbi.nlm.nih.gov/pubmed/35474094?tool=bestpractice.com
[38]Di Cola I, Di Muzio C, Conforti A, et al. Adult-onset Still's disease with elderly onset: results from a multicentre study. Clin Exp Rheumatol. 2022 Sep;40(8):1517-25.
https://www.doi.org/10.55563/clinexprheumatol/0215kv
http://www.ncbi.nlm.nih.gov/pubmed/35579097?tool=bestpractice.com
Risk factors
When taking a history, consider the presence of risk factors for AOSD, such as age and sex:
Multiple studies have found that women account for between 60% and 80% of people with AOSD.[8]Bogdan M, Nitsch-Osuch A, Samel-Kowalik P, et al. Adult-onset Still's disease in Poland - a nationwide population-based study. Ann Agric Environ Med. 2021 Jun 14;28(2):250-4.
https://www.doi.org/10.26444/aaem/132451
http://www.ncbi.nlm.nih.gov/pubmed/34184506?tool=bestpractice.com
[11]Lenert A, Oh G, Ombrello MJ, et al. Clinical characteristics and comorbidities in adult-onset Still's disease using a large US administrative claims database. Rheumatology (Oxford). 2020 Jul 1;59(7):1725-33.
https://www.doi.org/10.1093/rheumatology/kez622
http://www.ncbi.nlm.nih.gov/pubmed/31965185?tool=bestpractice.com
[12]Mehta BY, Ibrahim S, Briggs W, et al. Racial/ethnic variations in morbidity and mortality in adult onset Still's disease: an analysis of national dataset. Semin Arthritis Rheum. 2019 Dec;49(3):469-73.
https://www.doi.org/10.1016/j.semarthrit.2019.04.004
http://www.ncbi.nlm.nih.gov/pubmed/31109638?tool=bestpractice.com
[16]Balci MA, Pamuk ON, Pamuk GE, et al. AB1142 epidemiology and outcome of adult-onset Still’s disease in Northwestern Thrace region in Turkey. Ann Rheum Dis. 2015;74(suppl 2):1284.[17]Hocevar A, Rotar Z, Krosel M, et al. SAT0524 the incidence rate of adult onset Still's disease in Slovenia.Ann Rheum Dis. 2020;79 (suppl 1):1218-9.
https://ard.bmj.com/content/79/Suppl_1/1218
Most cases of AOSD occur in young adults, with a bimodal pattern showing two peaks of onset at ages 16-25 years and 36-46 years.[1]Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018 Sep;93:24-36.
https://www.doi.org/10.1016/j.jaut.2018.07.018
http://www.ncbi.nlm.nih.gov/pubmed/30077425?tool=bestpractice.com
[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
However, there is now increasing evidence of a further peak between the ages of 60 and 65 years.[7]Sfriso P, Priori R, Valesini G, et al. Adult-onset Still's disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016 Jul;35(7):1683-9.
https://www.doi.org/10.1007/s10067-016-3308-8
http://www.ncbi.nlm.nih.gov/pubmed/27207567?tool=bestpractice.com
[8]Bogdan M, Nitsch-Osuch A, Samel-Kowalik P, et al. Adult-onset Still's disease in Poland - a nationwide population-based study. Ann Agric Environ Med. 2021 Jun 14;28(2):250-4.
https://www.doi.org/10.26444/aaem/132451
http://www.ncbi.nlm.nih.gov/pubmed/34184506?tool=bestpractice.com
[9]Magadur-Joly G, Billaud E, Barrier JH, et al. Epidemiology of adult Still's disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995 Jul;54(7):587-90.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1009940
http://www.ncbi.nlm.nih.gov/pubmed/7668903?tool=bestpractice.com
[10]Evensen KJ, Nossent HC. Epidemiology and outcome of adult-onset Still's disease in Northern Norway. Scand J Rheumatol. 2006 Jan-Feb;35(1):48-51.
https://www.doi.org/10.1080/03009740510026616
http://www.ncbi.nlm.nih.gov/pubmed/16467042?tool=bestpractice.com
[16]Balci MA, Pamuk ON, Pamuk GE, et al. AB1142 epidemiology and outcome of adult-onset Still’s disease in Northwestern Thrace region in Turkey. Ann Rheum Dis. 2015;74(suppl 2):1284. Case series suggest that around 7% to 10% of cases are first diagnosed in patients older than 60 years of age although delayed diagnosis may be a contributory factor.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Ask about any preceding infection
It is likely AOSD is the manifestation of a dysregulated immune system, precipitated by a preceding viral or bacterial illness.[18]Wouters JM, van der Veen J, van de Putte LB, et al. Adult onset Still's disease and viral infections. Ann Rheum Dis. 1988 Sep;47(9):764-7.
https://www.doi.org/10.1136/ard.47.9.764
http://www.ncbi.nlm.nih.gov/pubmed/3178317?tool=bestpractice.com
[19]Escudero FJ, Len O, Falcó V, et al. Rubella infection in adult onset Still's disease. Ann Rheum Dis. 2000 Jun;59(6):493.
https://www.doi.org/10.1136/ard.59.6.490c
http://www.ncbi.nlm.nih.gov/pubmed/10885978?tool=bestpractice.com
[20]Jia J, Shi H, Liu M, et al. Cytomegalovirus infection may trigger adult-onset Still's disease onset or relapses. Front Immunol. 2019;10:898.
https://www.doi.org/10.3389/fimmu.2019.00898
http://www.ncbi.nlm.nih.gov/pubmed/31068953?tool=bestpractice.com
[21]Perez C, Artola V. Adult Still's disease associated with Mycoplasma pneumoniae infection. Clin Infect Dis. 2001 Mar 15;32(6):E105-6.
https://www.doi.org/10.1086/319342
http://www.ncbi.nlm.nih.gov/pubmed/11247732?tool=bestpractice.com
AOSD is thought to have a polygenic basis, whereby genetic factors predispose the patient to aberrant and sustained inflammatory responses in the context of an environmental or infectious trigger (e.g., cytomegalovirus, Epstein-Barr virus, rubella, Mycoplasma).[1]Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018 Sep;93:24-36.
https://www.doi.org/10.1016/j.jaut.2018.07.018
http://www.ncbi.nlm.nih.gov/pubmed/30077425?tool=bestpractice.com
[39]Hofheinz K, Schett G, Manger B. Adult onset Still's disease associated with malignancy - cause or coincidence? Semin Arthritis Rheum. 2016 Apr;45(5):621-6.
https://www.doi.org/10.1016/j.semarthrit.2015.10.003
http://www.ncbi.nlm.nih.gov/pubmed/26581485?tool=bestpractice.com
[40]Liozon E, Ly KH, Vidal-Cathala E, et al. Adult-onset Still's disease as a manifestation of malignancy: report of a patient with melanoma and literature review [in French]. Rev Med Interne. 2014 Jan;35(1):60-4.
https://www.doi.org/10.1016/j.revmed.2013.02.014
http://www.ncbi.nlm.nih.gov/pubmed/24094701?tool=bestpractice.com
[41]Gaggiano C, Rigante D, Vitale A, et al. Hints for genetic and clinical differentiation of adult-onset monogenic autoinflammatory diseases. Mediators Inflamm. 2019;2019:3293145.
https://www.doi.org/10.1155/2019/3293145
http://www.ncbi.nlm.nih.gov/pubmed/32082075?tool=bestpractice.com
Clinical course
When taking a history, be aware that the duration of symptoms and clinical course may vary between individual patients due to the different patterns of AOSD.
There are traditionally three broad patterns of disease:[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[42]Cush JJ, Medsger TA Jr, Christy WC, et al. Adult-onset Still's disease. Clinical course and outcome. Arthritis Rheum. 1987 Feb;30(2):186-94.
https://www.doi.org/10.1002/art.1780300209
http://www.ncbi.nlm.nih.gov/pubmed/3827959?tool=bestpractice.com
Monocyclic systemic (21% to 64% of cases): typically consists of one episode lasting a few months followed by remission.
Polycyclic systemic (9% to 50%): intermittent episodes or flares.
Chronic articular (12% to 56%): persisting symptoms, particularly affecting the joints.
More recently, a dichotomous classification system has been used, categorizing clinical AOSD subtypes as:[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Systemic (e.g., high fever, rash, multiorgan involvement) or
Articular (joint disease most prominent).
With future research, it is likely that these patterns will be further refined to incorporate clinical features, genetic analysis, cytokine profiles, and responses to treatment, as well as the manner in which the disease subtypes may evolve into separate autoinflammatory phenotypes as a consequence of prolonged immune dysregulation.[43]Mitrovic S, Fautrel B. Clinical phenotypes of adult-onset Still's disease: new insights from pathophysiology and literature findings. J Clin Med. 2021 Jun 15;10(12):2633.
https://www.doi.org/10.3390/jcm10122633
http://www.ncbi.nlm.nih.gov/pubmed/34203779?tool=bestpractice.com
Physical exam
Perform a full physical exam to identify the physical findings of AOSD and to exclude other conditions. This is particularly important as AOSD is typically a diagnosis of exclusion.
Common physical findings of AOSD include:[1]Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018 Sep;93:24-36.
https://www.doi.org/10.1016/j.jaut.2018.07.018
http://www.ncbi.nlm.nih.gov/pubmed/30077425?tool=bestpractice.com
[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[44]Nigrovic PA, Schneider R. Systemic juvenile idiopathic arthritis and adult onset still disease. In: Hashkes PJ, Laxer RM, Simon, A, eds. Textbook of autoinflammation. Cham, Switzerland: Springer Nature Switzerland AG; 2019, 587-616.
A high-spiking fever (as described above)
Inflammatory arthritis (as described above)
A characteristic skin rash (as described above)
Lymphadenopathy (often cervical) (frequency: 28% to 51%)
Splenomegaly (frequency: 25% to 43%).
Perform a targeted exam of symptomatic joints to identify inflammatory disease.
Less common findings include:[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
Hepatomegaly: studies vary on how frequently this occurs (7% to 71%).[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Signs of pericarditis (present in 3% to 17%) and myocarditis (in up to 7%).[7]Sfriso P, Priori R, Valesini G, et al. Adult-onset Still's disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016 Jul;35(7):1683-9.
https://www.doi.org/10.1007/s10067-016-3308-8
http://www.ncbi.nlm.nih.gov/pubmed/27207567?tool=bestpractice.com
[25]Hu QY, Zeng T, Sun CY, et al. Clinical features and current treatments of adult-onset Still's disease: a multicentre survey of 517 patients in China. Clin Exp Rheumatol. 2019 Nov-Dec;37(6 suppl 121):52-7.
https://www.clinexprheumatol.org/abstract.asp?a=13551
http://www.ncbi.nlm.nih.gov/pubmed/31573475?tool=bestpractice.com
[29]Asanuma YF, Mimura T, Tsuboi H, et al. Nationwide epidemiological survey of 169 patients with adult Still's disease in Japan. Mod Rheumatol. 2015 May;25(3):393-400.
https://www.doi.org/10.3109/14397595.2014.974881
http://www.ncbi.nlm.nih.gov/pubmed/25382730?tool=bestpractice.com
[30]Kalyoncu U, Solmaz D, Emmungil H, et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still's disease: data from a large multicenter cohort. J Autoimmun. 2016 May;69:59-63.
https://www.doi.org/10.1016/j.jaut.2016.02.010
http://www.ncbi.nlm.nih.gov/pubmed/26970681?tool=bestpractice.com
[45]Gracia-Ramos AE, Contreras-Ortíz JA. Myocarditis in adult-onset Still's disease: case-based review. Clin Rheumatol. 2020 Mar;39(3):933-47.
http://www.ncbi.nlm.nih.gov/pubmed/31745741?tool=bestpractice.com
These are associated with treatment resistance and a less favorable prognosis.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
Keep in mind important differentials to exclude, such as:
Vasculitis
Malignancy
Systemic lupus erythematosus
Rheumatoid arthritis
Mononeuritis multiplex (perform a thorough neurologic exam)
The sequelae of infectious endocarditis (perform a thorough cardiac exam).
See Differentials.
Complications of AOSD as presenting features
Consider the possibility that a patient may present with one or more complications of AOSD. Macrophage activation syndrome (MAS) is the most common and significant of these complications. Reports of its frequency vary widely, ranging from 1.7% to 23.0% of AOSD patients, and patients with an older onset (≥65 years) are three to five times more likely to develop it.[1]Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018 Sep;93:24-36.
https://www.doi.org/10.1016/j.jaut.2018.07.018
http://www.ncbi.nlm.nih.gov/pubmed/30077425?tool=bestpractice.com
[4]Macovei LA, Burlui A, Bratoiu I, et al. Adult-onset Still's disease-a complex disease, a challenging treatment. Int J Mol Sci. 2022 Oct 24;23(21):12810.
https://www.doi.org/10.3390/ijms232112810
http://www.ncbi.nlm.nih.gov/pubmed/36361602?tool=bestpractice.com
[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[6]Al-Hakim A, Mistry A, Savic S. Improving diagnosis and clinical management of acquired systemic autoinflammatory diseases. J Inflamm Res. 2022;15:5739-55.
https://www.doi.org/10.2147/JIR.S343261
http://www.ncbi.nlm.nih.gov/pubmed/36238769?tool=bestpractice.com
This highlights the need to consider the diagnosis of AOSD in those presenting with MAS and conversely to consider MAS in any acutely unwell patient with a preexisting diagnosis of AOSD.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
[7]Sfriso P, Priori R, Valesini G, et al. Adult-onset Still's disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016 Jul;35(7):1683-9.
https://www.doi.org/10.1007/s10067-016-3308-8
http://www.ncbi.nlm.nih.gov/pubmed/27207567?tool=bestpractice.com
[11]Lenert A, Oh G, Ombrello MJ, et al. Clinical characteristics and comorbidities in adult-onset Still's disease using a large US administrative claims database. Rheumatology (Oxford). 2020 Jul 1;59(7):1725-33.
https://www.doi.org/10.1093/rheumatology/kez622
http://www.ncbi.nlm.nih.gov/pubmed/31965185?tool=bestpractice.com
[16]Balci MA, Pamuk ON, Pamuk GE, et al. AB1142 epidemiology and outcome of adult-onset Still’s disease in Northwestern Thrace region in Turkey. Ann Rheum Dis. 2015;74(suppl 2):1284.[28]Zhang Y, Yang Y, Bai Y, et al. Clinical characteristics and follow-up analysis of adult-onset Still's disease complicated by hemophagocytic lymphohistiocytosis. Clin Rheumatol. 2016 May;35(5):1145-51.
https://www.doi.org/10.1007/s10067-016-3178-0
http://www.ncbi.nlm.nih.gov/pubmed/26809798?tool=bestpractice.com
Evaluation for MAS is advised if risk factors are present (e.g., high clinical disease activity and/or laboratory markers such as high serum ferritin and cytopenia [particularly leukopenia]).[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
In practice, it is not uncommon for a patient who has needed intensive care for MAS to be later managed for underlying AOSD. Seek an expert opinion where other rheumatologic and infectious causes have been ruled out.
Initial investigations
Exclude other more common causes of inflammation, as these can be fatal if left untreated or inappropriately treated. Investigations commonly start with those for a patient with a fever of unknown origin.[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
Request the following initial investigations:
Routine bloods such as CBC, renal panel, liver function tests, and C-reactive protein and/or erythrocyte sedimentation rate.
A septic screen, including carefully taken blood cultures and, where available, a procalcitonin to identify any underlying bacterial infections.
Radiologic investigations, such as a chest x-ray and renal/liver ultrasound scan as part of an initial septic screen. More extensive imaging may be done as differentials are increasingly ruled out.
If pericarditis is suspected, confirm with an ECG and echocardiogram.
If myocarditis is suspected, investigate with an ECG, cardiac enzymes, echocardiogram, and cardiac MRI.
Typical biochemical and hematologic findings include:[1]Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018 Sep;93:24-36.
https://www.doi.org/10.1016/j.jaut.2018.07.018
http://www.ncbi.nlm.nih.gov/pubmed/30077425?tool=bestpractice.com
[4]Macovei LA, Burlui A, Bratoiu I, et al. Adult-onset Still's disease-a complex disease, a challenging treatment. Int J Mol Sci. 2022 Oct 24;23(21):12810.
https://www.doi.org/10.3390/ijms232112810
http://www.ncbi.nlm.nih.gov/pubmed/36361602?tool=bestpractice.com
[5]Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021 Aug;51(4):858-74.
https://www.doi.org/10.1016/j.semarthrit.2021.06.004
http://www.ncbi.nlm.nih.gov/pubmed/34175791?tool=bestpractice.com
[46]Colafrancesco S, Priori R, Valesini G. Presentation and diagnosis of adult-onset Still's disease: the implications of current and emerging markers in overcoming the diagnostic challenge. Expert Rev Clin Immunol. 2015 Jun;11(6):749-61.
https://www.doi.org/10.1586/1744666X.2015.1037287
http://www.ncbi.nlm.nih.gov/pubmed/25948106?tool=bestpractice.com
[47]Mitrovic S, Fautrel B. New markers for adult-onset Still's disease. Joint Bone Spine. 2018 May;85(3):285-93.
https://www.doi.org/10.1016/j.jbspin.2017.05.011
http://www.ncbi.nlm.nih.gov/pubmed/28529117?tool=bestpractice.com
There may be elevated serum creatinine and/or abnormal LFTs, particularly elevations in aspartate and alanine aminotransferase.
Further investigations
Once AOSD is suspected, a large number of further investigations may be required to exclude the differential diagnoses and establish AOSD as the primary diagnosis. This may include extensive infectious screens including targeted cultures, interferon-gamma release assays (IGRAs) serology, and polymerase chain reaction (PCR) tests for specific infections; echocardiograms; lymph node biopsy; bone marrow biopsy; CT scans; and fluorodeoxyglucose (FDG)-positron emission tomography (PET) scans as more deep-seated infections, various malignancies, and MAS as a presenting complication need to be excluded.
Perform further tests to screen for autoimmune/rheumatologic conditions. These investigations include but are not limited to:
Rheumatoid factor (RF)
Anticitrullinated peptide (ACPA)
Antineutrophil cytoplasmic autoantibodies (ANCA)
Antinuclear antibodies (ANA)
HLA-B27
Muscle MRI/biopsy.
Check the ferritin level in any patient who has had differentials such as rheumatoid arthritis ruled out and who has or has had characteristic features of AOSD (e.g., persistent high temperatures and arthralgia, especially if supported by the characteristic rash or pharyngitis).[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
Hyperferritinemia is common (89%) in people with AOSD, though in isolation is poorly predictive of the disease.[47]Mitrovic S, Fautrel B. New markers for adult-onset Still's disease. Joint Bone Spine. 2018 May;85(3):285-93.
https://www.doi.org/10.1016/j.jbspin.2017.05.011
http://www.ncbi.nlm.nih.gov/pubmed/28529117?tool=bestpractice.com
However, the combination of a markedly elevated serum ferritin (≥5 × ULN) together with low fraction of glycosylated ferritin (<20%) can act as a sensitive and specific marker for AOSD.[1]Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018 Sep;93:24-36.
https://www.doi.org/10.1016/j.jaut.2018.07.018
http://www.ncbi.nlm.nih.gov/pubmed/30077425?tool=bestpractice.com
[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
[47]Mitrovic S, Fautrel B. New markers for adult-onset Still's disease. Joint Bone Spine. 2018 May;85(3):285-93.
https://www.doi.org/10.1016/j.jbspin.2017.05.011
http://www.ncbi.nlm.nih.gov/pubmed/28529117?tool=bestpractice.com
Diagnostic classification criteria
If ferritin is elevated, use diagnostic classification criteria for AOSD to help confirm the diagnosis.
A clinical diagnosis of AOSD is supported by fulfillment of the Yamaguchi criteria, developed in 1992.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
These are the most widely used and validated criteria for confirming a clinical diagnosis of AOSD, with a sensitivity of 96.2% and specificity of 92.1%.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
[4]Macovei LA, Burlui A, Bratoiu I, et al. Adult-onset Still's disease-a complex disease, a challenging treatment. Int J Mol Sci. 2022 Oct 24;23(21):12810.
https://www.doi.org/10.3390/ijms232112810
http://www.ncbi.nlm.nih.gov/pubmed/36361602?tool=bestpractice.com
[48]Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992 Mar;19(3):424-30.
http://www.ncbi.nlm.nih.gov/pubmed/1578458?tool=bestpractice.com
Yamaguchi criteria require the exclusion of infections, other rheumatic diseases, and malignancies.[47]Mitrovic S, Fautrel B. New markers for adult-onset Still's disease. Joint Bone Spine. 2018 May;85(3):285-93.
https://www.doi.org/10.1016/j.jbspin.2017.05.011
http://www.ncbi.nlm.nih.gov/pubmed/28529117?tool=bestpractice.com
If AOSD is suspected, check the glycosylated serum ferritin level if available as the Fautrel criteria can provide an invaluable tool in identifying the condition, ensuring early specialist care and management.[4]Macovei LA, Burlui A, Bratoiu I, et al. Adult-onset Still's disease-a complex disease, a challenging treatment. Int J Mol Sci. 2022 Oct 24;23(21):12810.
https://www.doi.org/10.3390/ijms232112810
http://www.ncbi.nlm.nih.gov/pubmed/36361602?tool=bestpractice.com
[6]Al-Hakim A, Mistry A, Savic S. Improving diagnosis and clinical management of acquired systemic autoinflammatory diseases. J Inflamm Res. 2022;15:5739-55.
https://www.doi.org/10.2147/JIR.S343261
http://www.ncbi.nlm.nih.gov/pubmed/36238769?tool=bestpractice.com
The Fautrel criteria were developed in 2002 and allow the process to be streamlined as they do not require any exclusion criteria.[49]Fautrel B, Zing E, Golmard JL, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002 May;81(3):194-200.
https://www.doi.org/10.1097/00005792-200205000-00003
http://www.ncbi.nlm.nih.gov/pubmed/11997716?tool=bestpractice.com
The Fautrel criteria include serum glycosylated ferritin (<20%) measurements, although this test may not be available in all locations.[2]Vordenbäumen S, Feist E, Rech J, et al. Diagnosis and treatment of adult-onset Still's disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(suppl 2):81-92.
https://www.doi.org/10.1007/s00393-022-01294-2
http://www.ncbi.nlm.nih.gov/pubmed/36520170?tool=bestpractice.com
[39]Hofheinz K, Schett G, Manger B. Adult onset Still's disease associated with malignancy - cause or coincidence? Semin Arthritis Rheum. 2016 Apr;45(5):621-6.
https://www.doi.org/10.1016/j.semarthrit.2015.10.003
http://www.ncbi.nlm.nih.gov/pubmed/26581485?tool=bestpractice.com
[49]Fautrel B, Zing E, Golmard JL, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002 May;81(3):194-200.
https://www.doi.org/10.1097/00005792-200205000-00003
http://www.ncbi.nlm.nih.gov/pubmed/11997716?tool=bestpractice.com
The Fautrel criteria have been validated to give a sensitivity of 96.3%, specificity of 98.9%, and positive and negative predictive values of 94.5% and 99.3%, respectively.[50]Lebrun D, Mestrallet S, Dehoux M, et al. Validation of the Fautrel classification criteria for adult-onset Still's disease. Semin Arthritis Rheum. 2018 Feb;47(4):578-85.
https://www.doi.org/10.1016/j.semarthrit.2017.07.005
http://www.ncbi.nlm.nih.gov/pubmed/28760536?tool=bestpractice.com
See Criteria for full details of the two classification criteria mentioned above.
Empirical corticosteroids
In practice, once infection, malignancy, and rheumatologic diseases are ruled out, a common form of investigation is "treat to test" with corticosteroids. A prompt response to corticosteroids would be seen in a person with AOSD and may guide the clinician when considering further investigations and more targeted therapies.
Genetic testing
Once initial testing is complete and an autoinflammatory condition is suspected, genetic analysis is increasingly playing a role in the identification of the specific underlying condition. Though there is no genetic test for AOSD, it is important to exclude known inherited and acquired monogenic disorders.[6]Al-Hakim A, Mistry A, Savic S. Improving diagnosis and clinical management of acquired systemic autoinflammatory diseases. J Inflamm Res. 2022;15:5739-55.
https://www.doi.org/10.2147/JIR.S343261
http://www.ncbi.nlm.nih.gov/pubmed/36238769?tool=bestpractice.com
A useful database of these conditions is available.
Infevers: The registry of hereditary auto-inflammatory disorders mutations
Opens in new window
In young adults, primary hemophagocytic lymphohistiocytosis should be excluded, while in adults, genetic analysis can reveal either inherited mutations or those acquired by chance in later life, known as genetic somatic mosaicism. An example of both can be found in the group of conditions known as NLRP3-autoinflammatory diseases (NLRP3-AID), where mutations in the NLRP3 gene lead to a spectrum of autoinflammatory diseases.[6]Al-Hakim A, Mistry A, Savic S. Improving diagnosis and clinical management of acquired systemic autoinflammatory diseases. J Inflamm Res. 2022;15:5739-55.
https://www.doi.org/10.2147/JIR.S343261
http://www.ncbi.nlm.nih.gov/pubmed/36238769?tool=bestpractice.com
In older adults presenting with autoinflammatory symptoms and cytopenias, acquired pathogenic mutations in the UBA1 gene should be sought to rule out VEXAS syndrome, which in the past has been commonly misdiagnosed as AOSD and has different treatment options.[6]Al-Hakim A, Mistry A, Savic S. Improving diagnosis and clinical management of acquired systemic autoinflammatory diseases. J Inflamm Res. 2022;15:5739-55.
https://www.doi.org/10.2147/JIR.S343261
http://www.ncbi.nlm.nih.gov/pubmed/36238769?tool=bestpractice.com
Emerging investigations
Each subtype of AOSD may have a characteristic cytokine profile, with interleukin (IL)-18/1β being predominant in the systemic subtype and IL-6 in the articular subtype.[43]Mitrovic S, Fautrel B. Clinical phenotypes of adult-onset Still's disease: new insights from pathophysiology and literature findings. J Clin Med. 2021 Jun 15;10(12):2633.
https://www.doi.org/10.3390/jcm10122633
http://www.ncbi.nlm.nih.gov/pubmed/34203779?tool=bestpractice.com
[51]Inoue N, Shimizu M, Tsunoda S, et al. Cytokine profile in adult-onset Still's disease: comparison with systemic juvenile idiopathic arthritis. Clin Immunol. 2016 Aug;169:8-13.
https://www.doi.org/10.1016/j.clim.2016.05.010
http://www.ncbi.nlm.nih.gov/pubmed/27263804?tool=bestpractice.com
[52]Jamilloux Y, Gerfaud-Valentin M, Martinon F, et al. Pathogenesis of adult-onset Still's disease: new insights from the juvenile counterpart. Immunol Res. 2015 Feb;61(1-2):53-62.
https://www.doi.org/10.1007/s12026-014-8561-9
http://www.ncbi.nlm.nih.gov/pubmed/25388963?tool=bestpractice.com
Cytokine testing may have a future role in risk stratification and identifying the most appropriate therapies. Risk of complications may also be predicted (e.g., increased likelihood of MAS with elevated IL-18).[51]Inoue N, Shimizu M, Tsunoda S, et al. Cytokine profile in adult-onset Still's disease: comparison with systemic juvenile idiopathic arthritis. Clin Immunol. 2016 Aug;169:8-13.
https://www.doi.org/10.1016/j.clim.2016.05.010
http://www.ncbi.nlm.nih.gov/pubmed/27263804?tool=bestpractice.com
[52]Jamilloux Y, Gerfaud-Valentin M, Martinon F, et al. Pathogenesis of adult-onset Still's disease: new insights from the juvenile counterpart. Immunol Res. 2015 Feb;61(1-2):53-62.
https://www.doi.org/10.1007/s12026-014-8561-9
http://www.ncbi.nlm.nih.gov/pubmed/25388963?tool=bestpractice.com
[53]Ahmed MG. Clinical significance of interleukin-18 and interleukin-6 on disease course of adult-onset still’s disease. Ann Rheum Dis, 2018;77(suppl 2):1162.
The proteins S100A12 and the S100A8/S100A9 dimer (calprotectin) are elevated in people with AOSD and have shown superiority over standard approaches in differentiating systemic juvenile idiopathic arthritis (sJIA) from other inflammatory conditions. They may, in the future, offer a more accurate biomarker for AOSD diagnosis and disease activity.[54]Holzinger D, Frosch M, Kastrup A, et al. The Toll-like receptor 4 agonist MRP8/14 protein complex is a sensitive indicator for disease activity and predicts relapses in systemic-onset juvenile idiopathic arthritis. Ann Rheum Dis. 2012 Jun;71(6):974-80.
https://www.doi.org/10.1136/annrheumdis-2011-200598
http://www.ncbi.nlm.nih.gov/pubmed/22267331?tool=bestpractice.com
[55]Aljaberi N, Tronconi E, Schulert G, et al. The use of S100 proteins testing in juvenile idiopathic arthritis and autoinflammatory diseases in a pediatric clinical setting: a retrospective analysis. Pediatr Rheumatol Online J. 2020 Jan 16;18(1):7.
https://www.doi.org/10.1186/s12969-020-0398-2
http://www.ncbi.nlm.nih.gov/pubmed/31948488?tool=bestpractice.com
[56]Kim HA, An JM, Nam JY, et al. Serum S100A8/A9, but not follistatin-like protein 1 and interleukin 18, may be a useful biomarker of disease activity in adult-onset Still's disease. J Rheumatol. 2012 Jul;39(7):1399-406.
https://www.doi.org/10.3899/jrheum.120079
http://www.ncbi.nlm.nih.gov/pubmed/22660800?tool=bestpractice.com
[57]Kim HA, Han JH, Kim WJ, et al. TLR4 endogenous ligand S100A8/A9 levels in adult-onset Still's disease and their association with disease activity and clinical manifestations. Int J Mol Sci. 2016 Aug 16;17(8):1342.
https://www.doi.org/10.3390/ijms17081342
http://www.ncbi.nlm.nih.gov/pubmed/27537874?tool=bestpractice.com
[58]Wittkowski H, et al. S100A12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheum. 2008 Dec;58(12):3924-31.[59]Bae CB, Suh CH, An JM, et al. Serum S100A12 may be a useful biomarker of disease activity in adult-onset Still's disease. J Rheumatol. 2014 Dec;41(12):2403-8.
https://www.doi.org/10.3899/jrheum.140651
http://www.ncbi.nlm.nih.gov/pubmed/25274901?tool=bestpractice.com
There is an association of certain HLA subtypes with AOSD. HLA-DQB1*06:02 (OR 2.70), HLA-DRB1*15:01 (OR 2.44), HLA-DRB1*11(OR 2.3), and HLA-DQA1*01:02 (OR 1.97) have all demonstrated an association with AOSD compared with healthy controls and may have a potential role in diagnosis in the future although further research is required.[60]Ombrello MJ, Remmers EF, Tachmazidou I, et al. HLA-DRB1*11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U S A. 2015 Dec 29;112(52):15970-5.
https://www.doi.org/10.1073/pnas.1520779112
http://www.ncbi.nlm.nih.gov/pubmed/26598658?tool=bestpractice.com
[61]Teng JL, Chen X, Chen J, et al. The amino acid variants in HLA II molecules explain the major association with adult-onset Still's disease in the Han Chinese population. J Autoimmun. 2021 Jan;116:102562.
https://www.doi.org/10.1016/j.jaut.2020.102562
http://www.ncbi.nlm.nih.gov/pubmed/33168359?tool=bestpractice.com