Aetiology
The majority of RCC occurs sporadically. Smoking is the most well-established risk factor.[27][30] Increasing age, obesity, and hypertension are associated with increased risk.[19][27][30] Renal transplantation, end-stage renal disease with dialysis, and exposure to pelvic radiation have been implicated.[26][31][32]
Several familial genetic syndromes have been identified.[30] Genes relating to kidney cancer include von Hippel-Lindau (VHL), folliculin (FLCN), succinate dehydrogenase (SDH), mesenchymal–epithelial transition factor (MET), fumarate hydratase (FH), and BAP1 (encodes for BRCA1 associated protein-1 [ubiquitin carboxy-terminal hydrolase]), which are inherited in an autosomal dominant manner.[33] Identification of genetic causes of RCC has implications for surveillance and use of targeted therapy.
VHL syndrome is associated with the clear cell variant of RCC. The lifetime risk of RCC in VHL syndrome is 70%.[33] Evidence suggests that 60% to 70% of patients with sporadic clear cell RCC also have VHL mutations.[34] Patients with VHL syndrome have a germline inactivation of one VHL allele (found on chromosome 3p); a second loss-of-function mutation is an early initiating event for VHL cancers such as RCC. VHL gene products act as tumour suppressors and are a key factor in clear cell RCC development. RCC will develop between the second and fourth decades of life in 25% to 45% of patients with VHL syndrome. The cancers are more often bilateral and/or multifocal; however, RCC in VHL syndrome usually has an indolent course, and tumours <3 cm in these patients rarely metastasise.[35] Regular surveillance for RCC in patients with VHL syndrome is warranted.[33]
Birt-Hogg-Dubé syndrome is caused by a germline mutation in the FLCN gene. It is predominantly associated with chromophobe and oncocytic renal cancers but does include other subtypes, including clear cell RCC. Patients with a FLCN mutation have an estimated risk of RCC of approximately 25%.[33]
Succinate dehydrogenase-related RCC has various histological types, including clear cell, and is associated with germline mutations in each of the SDH subunits, with SDHB being the most commonly associated gene.[33] The lifetime risk of RCC in SDHB mutation carriers is probably less than 10% to 15%.[33] Germline mutations in SDHB may present with a familial RCC-only phenotype, but mutations of the genes coding for the succinate dehydrogenase subunits are associated with pheochromocytomas and paragangliomas, and gastrointestinal stromal tumours.[33][36] Renal surveillance by magnetic resonance imaging can be combined with screening for phaeochromocytoma/paraganglioma in this group of patients.[33]
Other RCC histologies do not show a predominance of VHL mutations. For instance, type 1 papillary RCC mainly has mutations in c-MET, with type 2 (as part of hereditary leiomyomatosis and kidney cell cancer) having mutations in the FH gene.[33]
Tuberous sclerosis may be associated with early-onset RCC, but RCC is rare in this condition and renal lesions are most commonly angiomyolipomas.[33]
Pathophysiology
The various subtypes of RCC are associated with distinct cytogenetic abnormalities. Cancers with different genetic changes have different histology and clinical significance.[33][36]
Most RCCs have a clear cell histology, a minority have papillary or chromophobe histology:[37]
Clear cell carcinomas
Account for 70% to 90% of RCC.
Typically solitary cortical lesions, but bilateral (may also be multiple) cases are seen in von Hippel-Lindau syndrome.
Usually golden-yellow on gross pathology; may show cystic degeneration, haemorrhage, sarcomatoid change, and extension into renal vein; nucleoli grade is the most important prognostic factor after clinical stage.
Prognosis overall worse than papillary or chromophobe subtype, but response to systemic treatment higher; sporadic cases often show loss of chromosome 3p.
Von Hippel-Lindau (VHL) mutations seen in both sporadic cases and those associated with known VHL syndrome.
Papillary RCC
Account for 10% to 15% of RCC.
More commonly bilateral, or multifocal with variable degree of papillae.
Have frequent haemorrhage and necrosis.
Overall may have a better prognosis than clear cell carcinomas.
Associated with trisomy 7, 17, and loss of Y.
Chromophobe tumours
Account for 3% to 5% of RCC.
Large pale cells.
Usually relatively indolent disease course.
Knowledge of the molecular pathophysiology of clear cell RCC has been augmented by analysis of hereditary RCC syndromes such as VHL.[33][35] The VHL protein functions primarily as a ubiquitin ligase; one of its main degradation targets is hypoxia-inducible factor (HIF). HIF promotes the transcription of multiple hypoxia-related genes that are constitutively activated in the absence of VHL. These genes include vascular endothelial growth factor (VEGF), which promotes angiogenesis; platelet-derived growth factor (PDGF), which is a known oncogene; epidermal growth factor receptors, which promote cell growth; and matrix metalloproteinases, which are integral to tumour invasion. These HIF-controlled genes (especially VEGF) have been the targets of several molecular treatment strategies. Commonly used drugs in the treatment of RCC are tyrosine kinase inhibitors, designed to inhibit the VEGF and PDGF receptor pathways. Kinase proteins critical for angiogenesis are over-expressed or overactive in tumours. Inhibiting kinase function in tumour cells inhibits their interaction with the microenvironment, including the neovasculature, which has anti-cancer and toxic effects.[38] HIF transcription is itself controlled by the PI3 kinase pathway, of which the mammalian target of rapamycin (m-TOR) plays a key role in upregulation; m-TOR is also an RCC therapeutic target because of this. Other VHL cell targets include p53, certain RNA polymerases, and fibronectin. After the initiating loss of VHL allele(s), either as part of VHL syndromes or sporadically, accumulations of other non-random genetic defects are likely to contribute to tumour promotion.
The immune microenvironment is another targetable factor for cancer therapeutics. To prevent autoimmunity, the body has inherent checkpoint proteins, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 ligand 1 (PD-L1); however, these can be used by cancer cells to avoid detection by the immune system.[38] Many tumour cells express CTLA-4 and/or PD-L1, which binds to programmed cell death protein 1, and effectively inhibits T cells from attacking tumour cells.[38] Monoclonal antibodies directed against these cell death-related proteins prevent the activation of such immunosuppressive signals and overcome this immune tolerance, enabling an immunologically-driven anti-cancer effect.[38]
Tobacco exposure promotes the development of RCC.[39][40] Biological mechanisms may include oxidative stress and exposure to nitrosamines and other carcinogens in tobacco smoke. Nicotine has been shown to stimulate pathological angiogenesis and accelerate tumour growth.[30]
A few of the proposed mechanisms of obesity-induced RCC are increased lipid peroxidation forming DNA adducts, increased insulin-like growth factor, and higher glomerular filtration rate and nephrosclerosis causing increased carcinogen exposure.[26][30][41]
The proposed mechanism of hypertension-induced RCC is tubule damage and increased carcinogen exposure.[26][30]
Classification
Renal mass size
A small renal mass (SRM) is defined as a renal lesion less than 4 cm (or 3 cm by some authorities), which shows possible characteristics of an RCC (with abnormal enhancement) on imaging. SRMs (particularly those <2 cm) are more likely to be benign (up to 46% of those <1 cm, and 25% of those <2 cm).[3][4][5] Masses <3.5 cm (even if RCC) have low metastatic potential over 2-3 years. Masses that reach 4 cm and/or increase in size by 5 mm in 12 months may require intervention.[6]
TNM staging of RCC[7][8]
The TNM classification describes the extent of disease based on the following anatomic factors:
Size and extent of the primary tumour (T)
Regional lymph node involvement (N); and
Presence or absence of distant metastases (M).
Staging
Early-stage RCC (stages 1 and 2) is defined as tumour confined to kidney without regional lymph node or distant metastasis.
Stage 3 tumours extend into major veins, or invade the adrenal gland or perinephric tissue, but do not invade beyond the Gerota fascia. There may be metastasis in a single regional lymph node, but no evidence of distant metastasis.
Stage 4 tumours extend beyond the Gerota fascia or have distant metastasis.
World Health Organization (WHO) Classification: renal cell tumours[9]
The 2022 WHO classification introduced a molecular-driven renal tumour classification in addition to morphology-based renal tumours.[10]
Clear cell renal tumours
Clear cell renal cell carcinoma (RCC)
Multilocular cystic renal neoplasm of low malignant potential
Papillary renal tumours
Papillary adenoma
Papillary RCC (no longer divided into type I and II)
Oncocytic and chromophobe renal tumours
Oncocytoma of the kidney
Chromophobe RCC
Other oncocytic tumours of the kidney
Collecting duct tumours
Collecting duct carcinoma
Other renal tumours
Clear cell papillary renal cell tumour
Mucinous tubular and spindle cell carcinoma
Tubulocystic RCC
Acquired cystic disease-associated RCC
Eosinophilic solid and cystic (ESC) RCC
RCC NOS (not otherwise specified)
Molecularly defined renal tumours
TFE3-rearranged RCCs
TFEB-altered RCC (TFEB-rearranged RCC and TFEB amplified RCC)
ELOC (formerly TCEB1)-mutated RCC
Fumarate hydratase-deficient RCC
Succinate dehydrogenase-deficient RCC
ALK-rearranged RCCs
SMARCB1-deficient renal medullary carcinoma
World Health Organization (WHO)/International Society of Urological Pathology (ISUP): grading[11][12]
Nucleolar prominence defines grades 1-3 of clear cell and papillary RCCs. Extreme nuclear pleomorphism, or sarcomatoid and/or rhabdoid differentiation, defines grade 4 tumours.
Grade 1: tumour cell nucleoli absent or inconspicuous and basophilic at 400 times magnification
Grade 2: tumour cell nucleoli conspicuous and eosinophilic at 400 times magnification and visible, but not prominent at 100 times magnification
Grade 3: tumour cell nucleoli conspicuous and eosinophilic at 100 times magnification
Grade 4: tumours showing extreme nuclear pleomorphism, tumour giant cells, and/or the presence of any proportion of tumour showing sarcomatoid and/or rhabdoid de-differentiation.
Bosniak classification
The Bosniak classification was designed to classify cystic renal masses into five categories based on CT/MRI radiology.[13][14][15]
[Figure caption and citation for the preceding image starts]: Bosniak classificationTable created by Rodrigo R. Pessoa MD; used with permission [Citation ends].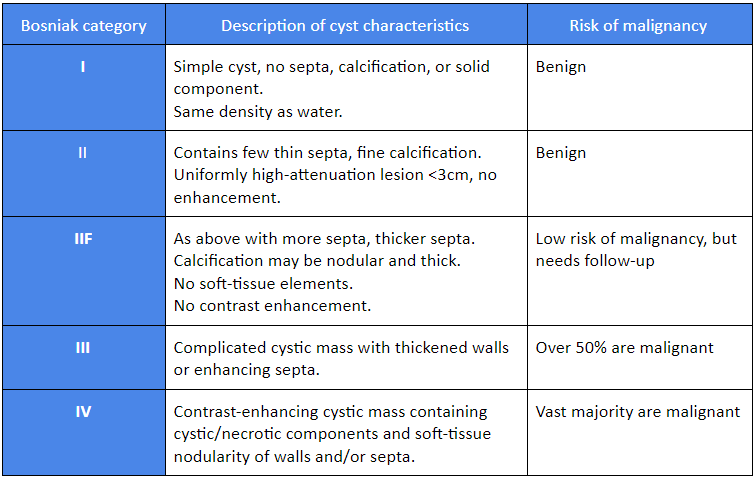
Use of this content is subject to our disclaimer