Etiology
RP is caused by mutations in genes that code for proteins important in the function and survival of photoreceptors and the retinal pigment epithelium. Mutations in more than 100 individual genes have been found to cause RP. The function of these different genes is diverse, including phototransduction, the retinoid cycle, photoreceptor structural integrity, phagocytosis of outer segment tips, structure and function of the cilium, metabolic homeostasis, mRNA splicing, and transcription of proteins. A family history of RP can help to reveal the pattern of inheritance.
RP is most frequently an isolated eye finding. However, there are some rare forms of syndromic RP, which present with systemic findings in addition to a typical pigmentary retinal degeneration. The most common forms of syndromic RP are Usher syndrome and Bardet-Biedl syndrome.[17] In syndromic forms of RP, the affected genes have an impact on photoreceptor function but additionally play an important role in other organ systems such as the ear or kidneys.
Syndromes associated with RP
Usher syndrome: this affects hearing and vision due to RP and a defective inner ear. Many patients also have balance problems, and children with this condition may be slow to walk.
Bardet-Biedl syndrome: there is usually both growth and intellectual impairment, with features including polydactyly or syndactyly, dilated cardiomyopathy, renal failure, and hepatic fibrosis. The degree of mental impairment can range from mild cognitive impairment to severe intellectual disability.
Alstrom syndrome: a rare autosomal recessive condition. Features include RP (cone-rod dystrophy), sensorineural hearing loss, infantile cardiomyopathy, and obesity. Without cardiomyopathy, the condition may not be diagnosed until the later development of diabetes mellitus.
Joubert syndrome: a rare condition usually inherited in an autosomal recessive manner that results in malformation of the cerebellar vermis (which connects the 2 halves of the cerebellum). Ataxia is a key feature, and others include learning difficulties, decreased muscle tone, renal cysts, sleep apnea, and extra fingers or toes.
Senior-Loken syndrome: a rare autosomal recessive disorder caused by a mutation in a gene involved in protein formation in the cilia. RP is accompanied by progressive kidney disease caused by defective nephron function and the formation of medullary cysts. Renal disease typically presents in the first year of life.
Neuronal ceroid-lipofuscinosis: a family of rare genetic neurodegenerative liposomal storage disorders. There is gradual motor and neurologic degeneration, seizures, and visual loss due to RP. Visual loss can be an early feature.
Kearns-Sayre syndrome: a mitochondrial disease that typically presents <20 years of age with visual loss due to RP. Other features may include ophthalmoplegia, hearing loss, ataxia, and dysphagia.
Bassen-Kornzweig disease (abetalipoproteinemia): a rare autosomal recessive condition that results in the poor absorption of fats, fat-soluble vitamins (A, D, E, K), and cholesterol. Symptoms usually present within the first few months of life and include failure to thrive, poor growth, and fat and blood in the stool. RP and ataxia may develop during later childhood.
Infantile Refsum disease: a metabolic disorder due to a defect in peroxisomal biogenesis. It usually presents in early childhood with visual and hearing impairment, poor muscle tone and coordination, intellectual impairment, and abnormal facial development.
Adult Refsum disease: this may be suspected in children presenting in late childhood with RP and combinations of anosmia, hearing loss, ataxia, cardiac arrhythmias, and short metacarpals and metatarsals. Some have a sensory or motor neuropathy.
Pathophysiology
While RP can be caused by mutations in an array of functionally different proteins, the underlying common pathophysiologic feature is apoptosis of the rod and cone photoreceptors. The cones are centrally arranged in the retina, and loss of cone photoreceptors will impair visual fields under light-adapted (photopic) conditions and can result in light sensitivity, decreased visual acuity, and loss of color vision. Loss of rod photoreceptors leads to impaired vision and constricted visual fields under dim (scotopic) conditions, as a well as problems with dark adaptation. Most commonly, the rod photoreceptors degenerate first, followed by the cones (rod-cone dystrophy), but certain genes can cause a cone-rod dystrophy, where the cones degenerate first followed by the rods. Finally, some mutations cause an isolated cone dystrophy.
As retinal photoreceptors degenerate, the retinal pigment epithelium migrates into the neural retinal layer to form bone spicules surrounding retinal vessels.[18] Secondary disorganization of the inner retinal layer is also seen.
Classification
Classification by inheritance
Autosomal dominant
X-linked
Autosomal recessive
Simplex: no family history, presumed to be recessive, but could be de novo dominant mutation as well
Multiplex: autosomal recessive with other affected siblings.
Mitochondrial (rare)
Digenic (rare)
Classification by functional loss
Rod-cone dystrophy
Cone-rod dystrophy
Cone dystrophy
Classification by age of onset
Leber congenital amaurosis: mutations in genes that cause severe disease with onset at <1 year of age
Juvenile RP: mutations in genes that cause severe disease with onset between 1 and 5 years of age
Typical RP: variable onset >5 years of age
Classification by pattern on fundus exam
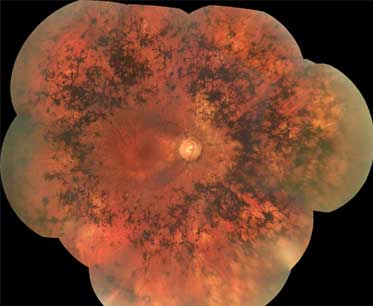
Classic pattern: starts in mid periphery and extends to the periphery and then centrally [Figure caption and citation for the preceding image starts]: Retinitis pigmentosaFrom the Oregon Retinal Degeneration Center collection [Citation ends].
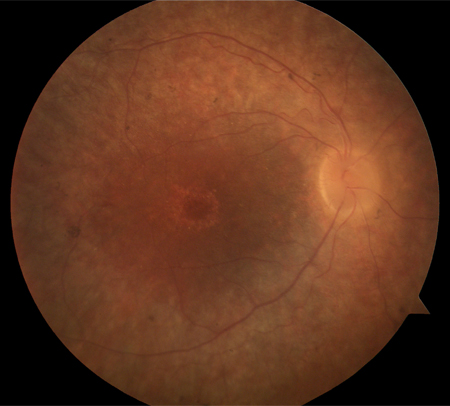
Bull's-eye maculopathy: seen in cone-rod and cone dystrophies (rare in typical RP) [Figure caption and citation for the preceding image starts]: Bull's-eye appearance in RPFrom the Oregon Retinal Degeneration Center collection [Citation ends].
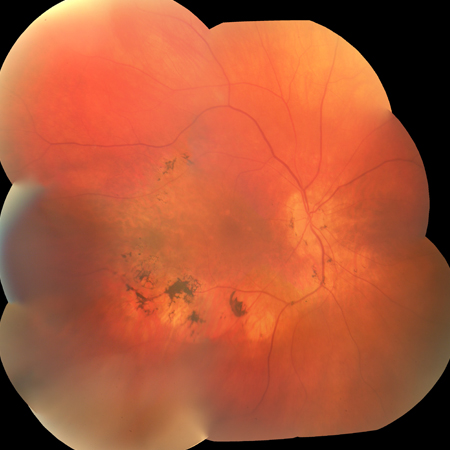
Sector RP: involving 1 or more regions of the retina in both eyes [Figure caption and citation for the preceding image starts]: Sector RPFrom the Oregon Retinal Degeneration Center collection [Citation ends].
Mosaic pattern: can be seen in female carriers of X-linked mutations
Pericentral RP: starts pericentrally within the arcades and extends centrally
Concentric: starts in the far periphery and progresses centrally
Unilateral: most likely not RP, but a disease that mimics RP
RP sine pigmento: lacks characteristic bone spicule pigmentation; may be seen in early disease
RP with preserved periarteriolar retinal pigment epithelium: classic pattern, but preserved retina near arterioles
Pigmented paravenous retinochoroidal atrophy: bone spicules limited to area around retinal veins.
Use of this content is subject to our disclaimer