Around 88% to 100% of patients with PJS will develop polyps.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Polyp-related symptoms usually arise in childhood and are seen by the age of 10 years in 33% and by 20 years in 50%.[5]Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul;59(7):975-86.
http://www.ncbi.nlm.nih.gov/pubmed/20581245?tool=bestpractice.com
Some patients present for a presymptomatic evaluation based on family history of the disease.
History and family history
Any history of intestinal polyposis, extraintestinal polyposis, mucocutaneous pigmentation, or malignancies in the patient and in the family should be noted. Gastric, small bowel, and colorectal polyposis occur in 88% to 100% of patients, with the majority appearing in the small bowel (60% to 90%) and colon (50% to 64%).[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Polyp growth begins in childhood by age 10 years, with most people experiencing symptoms such as bleeding, abdominal pain, intussusception, or obstruction by age 18 years.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Bleeding may be occult or overt and can result in iron-deficiency anemia and fatigue. Mucocutaneous pigmentation affects >95% of individuals and may often have been present in infancy but fades with time.[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
Physical exam
PJS is recognizable by the characteristic pigmented macules (flat, blue-gray to brown spots 1 to 5 millimeters in size) which occur most commonly in the perioral region and buccal mucosa (94%). They can also occur on the digits, palms, soles, face, forearms, perianal region, and rarely on the intestinal mucosa.[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
[15]Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006 Apr;4(4):408-15.
http://www.ncbi.nlm.nih.gov/pubmed/16616343?tool=bestpractice.com
The macules on the lips are distinctive as they cross the vermillion border and are typically darker and more densely clustered than freckles.[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
Pigmented macules on the skin increase in intensity from infancy and early childhood and often fade in adult life with complete disappearance in some patients. The buccal lesions usually persist.[13]Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndrome in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):442-52.
https://journals.lww.com/jpgn/Fulltext/2019/03000/Management_of_Peutz_Jeghers_Syndrome_in_Children.31.aspx
http://www.ncbi.nlm.nih.gov/pubmed/30585892?tool=bestpractice.com
Although uncommon, anemia may arise as a consequence of gastrointestinal bleeding secondary to polyps; pallor, fatigue, and tachycardia may be present.
Males should be examined for signs of testicular tumors. Mildly enlarged testicles (without masses) or features of feminizing precocious puberty (for example, bilateral gynecomastia) may suggest an underlying Sertoli cell tumor. The average age of diagnosis for testicular cancer is ages 9 years, with a range of 3 to 20 years.[16]Giardiello FM, Bresinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000 Dec;119(6):1447-53.
http://www.ncbi.nlm.nih.gov/pubmed/11113065?tool=bestpractice.com
Occasionally polyp prolapse per anus may be present in both sexes. [Figure caption and citation for the preceding image starts]: Characteristic pigmentation present on lips and buccal mucosaFrom the collection of Dr Carol A. Burke, used with permission [Citation ends].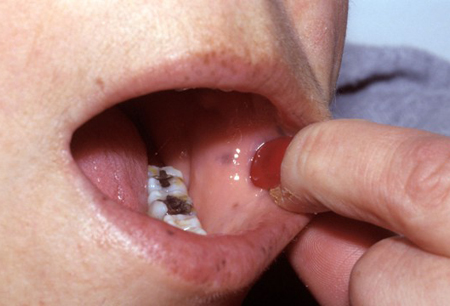
Diagnostic criteria
A diagnosis of PJS should be considered in a person who satisfies any one of the following:[5]Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul;59(7):975-86.
http://www.ncbi.nlm.nih.gov/pubmed/20581245?tool=bestpractice.com
Two or more histologically confirmed Peutz-Jeghers-type hamartomatous polyps (PJ-type polyps)
Any number of PJ-type polyps with a family history of PJS in a first-degree relative
Characteristic mucocutaneous pigmentation with a family history of PJS
Any number of PJ-type polyps and characteristic mucocutaneous pigmentation.
Individuals who meet the clinical criteria should undergo genetic evaluation.
Genetic testing
Genetic testing should be utilized to confirm the clinical diagnosis in those patients who satisfy the diagnostic criteria for PJS.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Sequencing of STK11 on chromosome 19p13.3, which encodes for the serine-threonine kinase LKB1, will detect 30% to 69% of mutations, and deletion/duplication analysis will find another 30% of germline alterations.[12]Amos CI, Keitheri-Cheteri MB, Sabripour M, et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004 May;41(5):327-33.
https://jmg.bmj.com/content/41/5/327.long
http://www.ncbi.nlm.nih.gov/pubmed/15121768?tool=bestpractice.com
[17]Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005 Dec;26(6):513-9.
http://www.ncbi.nlm.nih.gov/pubmed/16287113?tool=bestpractice.com
[18]Volikos E, Robinson J, Aittomaki K, et al. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J Med Genet. 2006 May;43(5):e18.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2564523
http://www.ncbi.nlm.nih.gov/pubmed/16648371?tool=bestpractice.com
A positive result confirms the diagnosis. Small and large deletions, insertions, splice site, and missense mutations in the STK11 gene have all been reported.[2]Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998 Jan;18(1):38-43.
http://www.ncbi.nlm.nih.gov/pubmed/9425897?tool=bestpractice.com
[3]Hemminki A, Makie D, Tomlinson I, et al. A serine threonine kinase gene defect in Peutz Jeghers syndrome. Nature. 1998 Jan 8;391(6663):184-7.
http://www.ncbi.nlm.nih.gov/pubmed/9428765?tool=bestpractice.com
A negative result significantly reduces the likelihood that the patient has PJS, although variant mutations may be missed. A variant of uncertain significance (VUS) could also be identified.
If an individual meets the clinical diagnostic criteria for PJS but has a negative or VUS result on genetic testing, then he or she should be managed as though they have PJS or evaluated further for another genetic syndrome.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Do not repeat a genetic test unless there is uncertainty about an existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[19]American College of Medical Genetics and Genomics. Five things physicians and patients should question. Choosing Wisely, an initiative of the ABIM Foundation. 2021 [internet publication].
https://web.archive.org/web/20230326143738/https://www.choosingwisely.org/societies/american-college-of-medical-genetics-and-genomics
Once a mutation is identified in a proband, at-risk family members can be tested for this family-specific alteration. It should be recognized that, while most patients will have an affected parent, approximately 25% of patients have de novo mutations.[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
Parents of patients with presumptive de novo mutations should be carefully evaluated for features of PJS (colonic polyps or mucocutaneous pigmentation). If one of the parents is affected, then testing should be offered to the siblings of the proband.
Whether inherited or de novo, all children of the proband have a 50% risk of inheriting the mutation and should be tested accordingly. After appropriate genetic counseling and informed consent, testing for at-risk family members may be performed during childhood.
Gene reviews: Peutz-Jeghers syndrome
Opens in new window
Endoscopic evaluation
Endoscopic evaluation is indicated for three groups of individuals:
Asymptomatic individuals with PJS undergoing surveillance
Symptomatic individuals or patients with laboratory or radiographic abnormalities suggestive of PJS
At-risk first-degree relatives who meet the phenotypic criteria (i.e., colonic polyps or mucocutaneous pigmentation) when the family mutation is not identified.
Surveillance of the colon, stomach, and duodenum by esophagogastroduodenoscopy (EGD) and colonoscopy should begin by age 8 years in affected asymptomatic individuals (and earlier in those with symptoms).[13]Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndrome in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):442-52.
https://journals.lww.com/jpgn/Fulltext/2019/03000/Management_of_Peutz_Jeghers_Syndrome_in_Children.31.aspx
http://www.ncbi.nlm.nih.gov/pubmed/30585892?tool=bestpractice.com
[20]Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020 Mar;69(3):411-44.
https://gut.bmj.com/content/69/3/411.long
http://www.ncbi.nlm.nih.gov/pubmed/31780574?tool=bestpractice.com
Small bowel surveillance with video capsule endoscopy (VCE) or magnetic resonance enterography (MRE) should also begin by ages 8 years.[5]Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul;59(7):975-86.
http://www.ncbi.nlm.nih.gov/pubmed/20581245?tool=bestpractice.com
[13]Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndrome in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):442-52.
https://journals.lww.com/jpgn/Fulltext/2019/03000/Management_of_Peutz_Jeghers_Syndrome_in_Children.31.aspx
http://www.ncbi.nlm.nih.gov/pubmed/30585892?tool=bestpractice.com
[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
[20]Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020 Mar;69(3):411-44.
https://gut.bmj.com/content/69/3/411.long
http://www.ncbi.nlm.nih.gov/pubmed/31780574?tool=bestpractice.com
[21]van Leerdam ME, Roos VH, van Hooft JE, et al. Endoscopic management of polyposis syndromes: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy. 2019 Sep;51(9):877-95.
https://www.thieme-connect.com/products/ejournals/html/10.1055/a-0965-0605
http://www.ncbi.nlm.nih.gov/pubmed/31342472?tool=bestpractice.com
Capsule endoscopy is safe to use in individuals with PJS and small bowel polyposis who lack obstructive symptoms. If a concern for capsule retention is present, a patency capsule should be utilized. In general, adverse events associated with diagnostic EGD are rare (reported rates: infection <0.3%, bleeding <0.1%, cardiopulmonary events <0.1%, perforation <0.01%).[22]ASGE Standards of Practice Committee, Coelho-Prabhu N, Forbes N, et al. Adverse events associated with EGD and EGD-related techniques. Gastrointest Endosc. 2022 Sep;96(3):389-401.e1.
https://www.giejournal.org/article/S0016-5107(22)00337-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35843754?tool=bestpractice.com
The most common endoscopic finding is diffuse hamartomatous polyposis. Gastric, small bowel, and colorectal polyposis occur in 88% to 100% of patients, with the majority appearing in the small bowel (60% to 90%) and colon (50% to 64%).[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
In rare cases, polyps have also occurred in the renal pelvis, urinary bladder, lungs, and nares. Polyps may be small or large, sessile, or pedunculated. They often have a characteristic cerebriform appearance due to smooth muscle arborization within the polyps. Multiple polyps are typical; usually the total number is below 20. Individual polyps vary in size from a few millimeters to several centimeters. Pathologically the polyp is hamartomatous in nature; smooth muscle core is covered by lamina propria and mature glandular epithelium. [Figure caption and citation for the preceding image starts]: Small bowel hamartomatous polyp identified on capsule endoscopyFrom the collection of Dr Carol A. Burke, used with permission [Citation ends].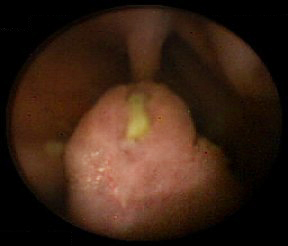 [Figure caption and citation for the preceding image starts]: Polypoid mass in the small bowelFrom the collection of Dr James Church, used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Polypoid mass in the small bowelFrom the collection of Dr James Church, used with permission [Citation ends].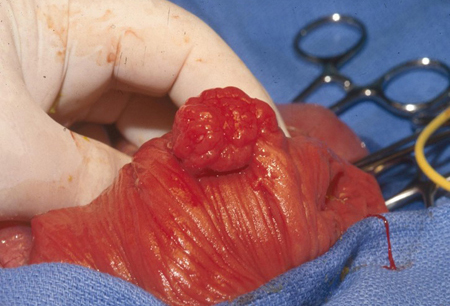 [Figure caption and citation for the preceding image starts]: Characteristic serosal dimpling resulting from a pedunculated hamartoma in the small bowel of a patient with PJSFrom the collection of Dr James Church, used with permission [Citation ends].
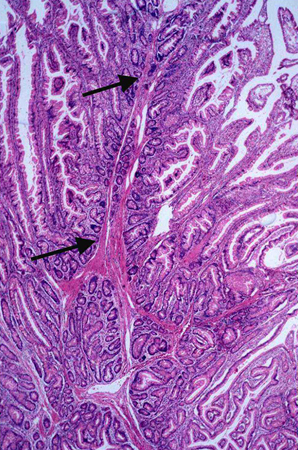
[Figure caption and citation for the preceding image starts]: Characteristic serosal dimpling resulting from a pedunculated hamartoma in the small bowel of a patient with PJSFrom the collection of Dr James Church, used with permission [Citation ends]. [Figure caption and citation for the preceding image starts]: Histology of a hamartomatous polypFrom the collection of Dr Carol A. Burke, used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Histology of a hamartomatous polypFrom the collection of Dr Carol A. Burke, used with permission [Citation ends].