Approach
The majority of patients with a temporary or functional delay progress in puberty spontaneously and achieve an adequate growth potential. However, patients with an organic defect, such as those with congenital gonadotropin deficiency or gonadal abnormalities, need early treatment to achieve an optimal final height and secondary sexual characteristics. In the absence of pubertal changes, height velocity declines from 5 cm (2 inches) per year at age 12 years by 1 cm (0.4 inches) per year for every year thereafter that puberty is not manifest. Patients should be assessed by a pediatric endocrinologist experienced in the diagnosis and treatment of delayed puberty.
History
The majority of those who present with delayed puberty are boys. Many patients seek medical help because of slow growth rather than slow pubertal development, as well as due to concerns that puberty has not started.
Assessment should initially include a detailed history for causes of a temporary delay. Any chronic medical condition can lead to delayed puberty, with or without short stature, and poor weight gain. Common conditions include malnutrition, chronic heart disease, moderate to severe asthma, cystic fibrosis, celiac disease, inflammatory bowel disease (Crohn disease and ulcerative colitis), inflammatory disorders (e.g., juvenile idiopathic arthritis), chronic renal failure, any chronic malignancy, and poorly controlled diabetes mellitus. A history of abnormal eating patterns or recent weight loss in girls must be sought. This may sometimes be associated with intense exercise routines and should prompt suspicion of an eating disorder such as anorexia nervosa or bulimia nervosa.
Constitutional delay - also known as constitutional delay of growth and puberty (CDGP), self-limited delayed puberty, or isolated delayed puberty - is the most common cause of delayed puberty in both males and females.[1][20][37] It tends to occur within families and is associated with short stature. A family history should be recorded, including heights of both parents and their ages at onset of pubertal development, as well as maternal age of menarche.[37]
History may reveal previous surgery, or radiation therapy to the brain (resulting in gonadotropin deficiency) or the abdomen or pelvis (gonadal damage) due to prior malignancy. Several chemotherapeutic agents can result in delayed puberty. Testicular pain or recent viral infections such as mumps may be indicative of testicular infection or torsion. Histiocytosis, sickle cell disease, and iron overload (associated with transfusion) can result in permanent gonadotropin deficiency.
There may be symptoms suggestive of other hormone deficits, such as growth failure due to growth hormone deficiency or symptoms of hypothyroidism, which may indicate a central (hypothalamo-pituitary) cause. This may be associated with a loss of smell (anosmia) suggestive of Kallmann syndrome.
Multiple endocrine gland abnormalities such as delayed puberty in association with diabetes mellitus and/or Addison disease should raise a suspicion of an autoimmune disorder. Rarely, congenital gonadotropin deficiency may be associated with congenital adrenal hypoplasia, and the history may be indicative of salt loss or adrenal crisis. The diagnosis may already be established, in which case the patient may be receiving glucocorticoid and mineralocorticoid treatment.[Figure caption and citation for the preceding image starts]: Growth in a patient with constitutional delay in growth and puberty; X indicates bone age; arrow indicates age at onset of pubertal developmentFrom the collection of Dr A. Mehta [Citation ends].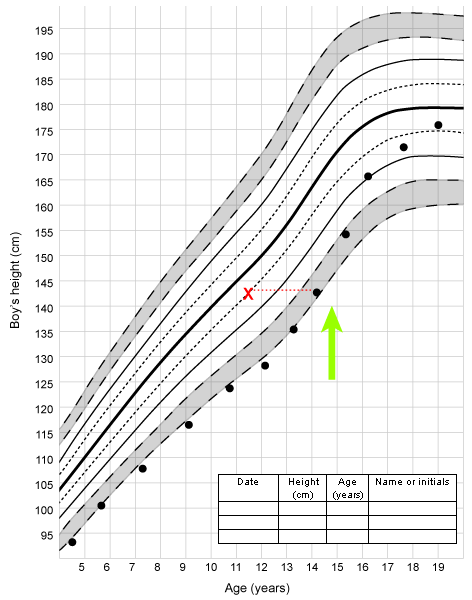
Tanner staging
Pubertal development and progress are best assessed by Tanner staging.[38][39]
The first sign of puberty in boys is an increase in the size of the testes. This is followed by penile and scrotal changes. Testicular size is documented as a measurement of the longest axis or by the testicular volume using the Prader orchidometer.
[Figure caption and citation for the preceding image starts]: Prader orchidometerCreated by BMJ Knowledge Centre [Citation ends].
 [Figure caption and citation for the preceding image starts]: Method of comparing testicular size using the Prader orchidometerFrom the collection of Dr A. Mehta; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Method of comparing testicular size using the Prader orchidometerFrom the collection of Dr A. Mehta; used with permission [Citation ends].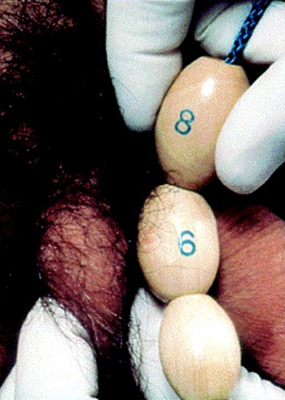
Volume of 4 mL or a longitudinal length of 2.5 cm (1 inch) defines the onset of puberty.[39] Growth of axillary hair and other changes such as that of the voice and an increase in growth velocity only occur in mid- to late puberty. Facial hair does not appear until late puberty.
The first demonstrable sign of puberty in girls is breast development. Caution must be exercised in exam of breast tissue in obese girls, as simple fat may be mistaken for breast tissue. Pubic and axillary hair, acne, and body odor develop as a result of androgens secreted from the adrenal gland. The peak growth spurt occurs in Tanner stage 3 breast development, and menarche occurs typically alongside Tanner stage 4 breast development. [Figure caption and citation for the preceding image starts]: Tanner staging: A, genital rating standards in boys; B, pubic hair rating standards in boys; C, breast rating standards in girls; D, pubic hair rating standards in girlsAdapted from Marshall WA, Tanner JM. Arch Dis Child. 1970;45:13-23; Marshall WA, Tanner JM. Arch Dis Child. 1969;44:291-303 [Citation ends].

Exam
Present and previous height and weight measurements should be accurately recorded. The majority of patients who present with delayed puberty are boys with constitutional delay who have a normal exam apart from short stature and a growth velocity just below the mean for age and sex. Those with a temporary delay such as with chronic illness may have the typical abnormalities of that disorder. It is important to assess the patient for features of malnutrition.
The patient should be assessed for body disproportion. Boys with Klinefelter syndrome have tall stature with a greater lower limb length. In addition, they may have dysgenetic testes (small and soft on palpation) and developmental delay, and they may have gynecomastia.[27]
Girls with Turner syndrome are typically short, with several dysmorphic features that include a low posterior hairline, a webbed neck, and prominent posteriorly rotated ears. Presenting features in many cases may be more subtle, and investigations to rule out Turner syndrome should be undertaken in any girl with a delay in puberty.[40][41] See Turner syndrome.
There may be other features to suggest a syndromic diagnosis (e.g., Prader-Willi, Bardet-Biedl, CHARGE, or septo-optic dysplasia).
A detailed central nervous system exam should be carried out to look for any focal deficits suggestive of an intracranial tumor. The sense of smell should be ascertained; anosmia suggests Kallmann syndrome, which is an association of organic hypogonadotropic hypogonadism and hypoplastic olfactory nerves.[21] Some patients with Kallmann syndrome have a genetic defect in genes such as ANOS1, FGF8, FGFR1, PROK2, or PROKR2.
[Figure caption and citation for the preceding image starts]: Evaluation of a patient with delayed puberty. CDGP, constitutional delay of growth and puberty; DP, delayed puberty; FT4, free T4; GHD, GH deficiency; PRL, prolactin; TFT, thyroid function testEndocr Rev. 2019 Oct; 40(5): 1285–1317; used with permission [Citation ends].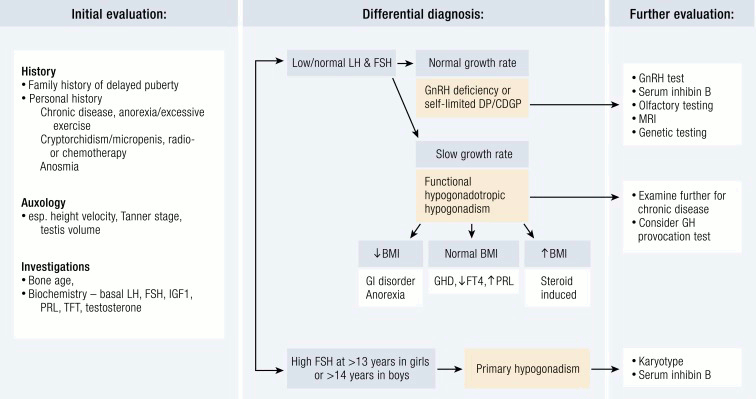
Initial investigations
Initial investigations should include a nondominant (typically, left) wrist radiograph to estimate bone age. The bone age indicates if constitutional delay is present or absent, and helps predict the estimated adult height range and its relation to the midparental height.[1] The appearance of representative epiphyseal centers on the x-ray is compared with age- and sex-appropriate published standards. The most commonly used method is that of Greulich and Pyle.[42]
Laboratory measurement of serum follicle-stimulating hormone (FSH) and luteinizing hormone (LH) concentrations will help differentiate patients with hypogonadotropic hypogonadism (low or normal levels) and hypergonadotropic hypogonadism (elevated levels).[1] The measurements should be performed on an early-morning blood sample using an ultrasensitive pediatric assay. As the hypothalamo-pituitary-gonadal axis is quiescent up to about 10 to 12 years of age, levels of these hormones in children <12 years of age must be interpreted with caution. It may be difficult to distinguish constitutional delay from organic gonadotropin deficiency, as the basal gonadotropin concentrations may be low or normal in both groups.
Further investigations and management depend on history, the severity of delay, height, and results of basal gonadotropin measurement.
Subsequent tests
Gonadotropin-releasing hormone (GnRH) testing
Can be considered in all patients with low basal gonadotropins; however, sensitivity and specificity is limited.[43] Girls with Turner syndrome and other patients with gonadal abnormalities typically have marked elevations in basal serum FSH and LH concentrations (in Turner syndrome from 8 to 9 years of age), and the GnRH test is most often not indicated in these patients.[44]
LH-releasing hormone (LHRH) is used to stimulate gonadotropins. It has been studied extensively in patients with low basal gonadotropins but does not always help differentiate between constitutional delay and organic gonadotropin deficiency, as stimulated serum FSH and LH concentrations following LHRH may be low in both groups, or may be normal and thus falsely reassuring in an individual with hypothalamic GnRH deficiency but preserved pituitary function.[45] Thus, it does not clarify whether an individual will progress in puberty spontaneously or has a permanent defect. Some patients with a temporary delay may therefore need to be treated with sex steroids in order to induce puberty, but when re-evaluated, after completion of growth and puberty, with a trial off treatment, may reveal normal serum FSH and LH concentrations.
Inhibin B and anti-Mullerian hormone
Basal inhibin B is a promising biochemical investigation for the diagnosis of delayed puberty. In males, it is produced exclusively by the Sertoli cells of the testes, and in females by the ovarian granulosa cells and the placenta. Low inhibin B of less than 35 pg/mL in prepubertal boys is a sensitive marker to discriminate permanent hypogonadotropic hypogonadism from constitutional or self‐limited delayed puberty.[46] In males, the trio of small testes volume (cut-off: 1.1 mL), low maximal stimulated LH on GnRH stimulation testing (cut-off: 4.3 IU/L), and low basal inhibin B concentration (cut-off: 61 pg/mL) have been proposed as the most effective discriminator between these two conditions.[18] In males with hypergonadotropic hypogonadism, low serum inhibin B reflects primary germ cell failure. The utility of inhibin B has been less clearly demonstrated in girls.
Anti-Mullerian hormone (AMH) is produced in males from the testicular Sertoli cells from the time of testicular differentiation to puberty. In females, it is secreted by the ovarian granulosa cells from birth until menopause, although the concentrations in girls are significantly lower. Patients with dysgenetic gonads have low serum AMH. Undetectable AMH and inhibin B are characteristic of congenital anorchia, but may also be seen in males and females with severe hypogonadotropic hypogonadism. AMH and inhibin B may be used, therefore, during assessment of testicular or ovarian function and capacity (as a marker of potential for fertility), and as a diagnostic tool in conjunction with serum gonadotropins for both hypo- and hypergonadotropic hypogonadism.[47]
Human chorionic gonadotropin (hCG) stimulation test
The hCG test is used to stimulate testicular production of testosterone, and has been suggested to be useful in some patients when combined with the LHRH test to help distinguish constitutional delay from hypogonadotropic hypogonadism.[45]
A rise in serum testosterone concentration is observed in constitutional delay; a decreased testosterone response is seen with hypogonadotropic hypogonadism.
Overnight sampling of gonadotropins
May demonstrate pulsatility, but its role in the long-term prognosis of delay is unclear. It is primarily undertaken in a research setting.
Other tests
Sex steroids: measurement of sex steroids is only useful in the context of hCG stimulation tests. Baseline measurement of serum sex steroids (testosterone and estradiol) are rarely diagnostic.
Chromosomal analysis: may reveal Klinefelter syndrome (47XXY) in boys or Turner syndrome (45X) in girls. The diagnosis of Turner syndrome (45X) should be considered in all short girls, with or without other phenotypic features suggestive of Turner syndrome.[41] Depending on the mosaicism, 45X/46XY mixed gonadal dysgenesis may present with ambiguous genitalia at birth or just with delayed puberty in adolescence.[40] Chromosomal microarray may be indicated in patients with hypergonadotropic hypogonadism with additional congenital abnormalities or syndromic features.[48]
Genetic sequencing: for individuals with suspected congenital hypogonadotropic hypogonadism or Kallmann syndrome, next-generation sequencing panel testing is now available in the UK and some other world regions.[49]
Imaging: pelvic ultrasound exam should be considered in girls with Turner syndrome or other forms of gonadal dysgenesis, as well as in girls with hypogonadotropic hypogonadism. In individuals with hypergonadotropic hypogonadism, it may reveal dysgenetic gonads. In those with hypogonadotropic hypogonadism, it is useful for the assessment of uterine and ovarian maturity (volume, shape, and endometrial thickness) for diagnosis and before starting treatment to allow monitoring of response to therapy. Abdominal ultrasound may reveal renal abnormalities such as agenesis or a horseshoe kidney. Echocardiography in girls with Turner syndrome may show cardiac defects including coarctation of the aorta and a bicuspid aortic valve. Magnetic resonance imaging of the brain should be considered if basal gonadotropins are low in the absence of a family history of delayed puberty. It is mandatory if other pituitary hormone levels are found to be deficient. Neuroimaging of the brain helps identify structural hypothalamo-pituitary abnormalities, midline brain defects, olfactory hypoplasia, and pituitary tumors.[1][50]
Other pituitary hormone tests to rule out combined pituitary deficiencies should be considered in patients with confirmed hypogonadotropic hypogonadism.
Measurement of serum ovarian antibodies to identify an autoimmune process.
Thyroid function tests and serum prolactin concentrations are useful to identify patients with hypothyroidism or hyperprolactinemia, both of which can present with delayed puberty.
To assess GnRH production by the hypothalamus, LH measurement in response to stimulation with kisspeptin has been proposed as a useful test to identify individuals with GnRH deficiency and thus permanent hypogonadotropic hypogonadism. Kisspeptin stimulates GnRH pulsatility, thus promoting LH - and to a lesser extent FSH - secretion. One clinical study found that maximal LH rise after kisspeptin administration was more accurate for the diagnosis of men with GnRH deficiency than GnRH stimulation testing.[51] In one parallel study in adolescents with pubertal delay (3 females and 13 males), peak LH post kisspeptin stimulation was demonstrated to be superior to GnRH stimulation testing for predicting capacity to progress through puberty.[52] While further research is required to delineate the parameters of using kisspeptin in clinical pediatric practice, this is a promising area for the biochemical diagnosis of GnRH deficiency in adolescence.
Use of this content is subject to our disclaimer