Etiology
The common etiologic denominator in ARLD is chronic, heavy alcohol ingestion. Alcohol is a risk factor for advanced liver disease and cirrhosis; however, the threshold of consumption at which risk emerges is unclear.[1][12] One systematic review found evidence of increased risk of mortality from cirrhosis among men and women drinking >12 to 24 g and >0 to 12 g of ethanol per day, respectively.[13]
Alcohol misuse or alcohol dependence alone are not suggestive of clinically important ARLD. Only about 10% to 20% of chronic heavy drinkers develop severe forms such as alcohol-related hepatitis or cirrhosis.[1][14][15] In patients with coexistent liver diseases related to hepatitis C or obesity, alcohol-related liver damage can occur with much lower alcohol consumption.[1][16][17] The risk of ARLD is at least 2 times higher in patients who are overweight.[18][19] Smoking is common among heavy drinkers; smoking not only accelerates the progression of fibrosis in patients with ARLD, but also encourages the development of hepatocellular carcinoma in patients with alcohol-related cirrhosis.[20][21] These behaviors are modifiable, and afford an opportunity to significantly alter the adverse impact of ARLD.
Meta-analyses of observational data indicate that a number of cytokine and alcohol-metabolizing enzyme gene polymorphisms may be associated with increased risk for ARLD:[22][23][24]
Tumor necrosis factor-alpha 238G>A polymorphism
Interleukin-6 gene polymorphisms (G [rs1800795], C [rs1800796] or G [rs1800797] allele or genotypes)
Patatin-like phospholipase domain protein 3 (PNPLA3) gene polymorphism (rs738409 C>G).
Associations may vary with ethnicity. Further, more rigorous research is required to delineate the relationship between specific polymorphisms and risk for ARLD.
Pathophysiology
Alcohol is metabolized mainly in the liver, through two main pathways: alcohol dehydrogenase and cytochrome P-450 2E1.[25][26]
Alcohol dehydrogenase is a hepatic enzyme that converts alcohol to acetaldehyde, which is subsequently metabolized to acetate by the mitochondrial enzyme acetaldehyde dehydrogenase. Alcohol dehydrogenase and acetaldehyde dehydrogenase reduce nicotinamide adenine dinucleotide (NAD) to NADH (reduced form of NAD). The altered ratio of NAD to NADH inhibits gluconeogenesis and increases fatty acid oxidation, which in turn promotes fatty infiltration in the liver.[26]
The cytochrome P-450 2E1 pathway generates free radicals through the oxidation of NADPH (reduced form of nicotinamide adenine dinucleotide phosphate [NADP]) to NADP. Chronic alcohol use upregulates cytochrome P-450 2E1 and produces more free radicals.[26]
Chronic alcohol exposure also activates a third site of metabolism: hepatic macrophages, which produce tumor necrosis factor (TNF)-alpha and induce the production of reactive oxygen species in the mitochondria.
People with alcohol use disorder are usually deficient in antioxidants, such as glutathione and vitamin E. Therefore, oxidative stress promotes hepatocyte necrosis and apoptosis in these patients. Free radicals can also induce lipid peroxidation, which can cause inflammation and fibrosis.[26] The alcohol metabolite acetaldehyde, when bound to cellular protein, produces antigenic adducts and induces inflammation. Alcohol also affects the barrier function of intestinal mucosa, producing endotoxemia, which leads to hepatic inflammation.[27]
Other pathways may have a role in the pathogenesis of ARLD.[28][Figure caption and citation for the preceding image starts]: Liver biopsy showing the typical histologic changes of alcohol-related steatosis (fatty liver)From the collection of Dr McClain; used with permission [Citation ends].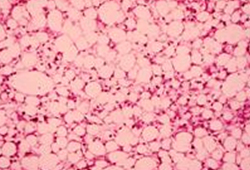 [Figure caption and citation for the preceding image starts]: Liver biopsy showing the typical histologic changes of alcohol-related steatohepatitisFrom the collection of Dr McClain; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Liver biopsy showing the typical histologic changes of alcohol-related steatohepatitisFrom the collection of Dr McClain; used with permission [Citation ends].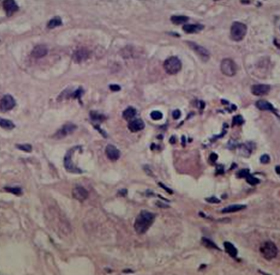 [Figure caption and citation for the preceding image starts]: Liver biopsy showing alcohol-related cirrhosisFrom the collection of Dr McClain; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Liver biopsy showing alcohol-related cirrhosisFrom the collection of Dr McClain; used with permission [Citation ends].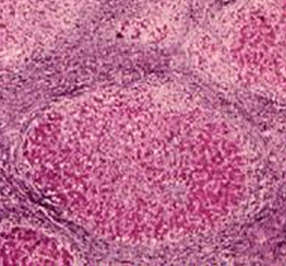
Use of this content is subject to our disclaimer