Etiology
Any disorder that results in hypocalcemia will elevate parathyroid hormone (PTH) levels and can serve as a cause of secondary hyperparathyroidism (SHPT).[9]
The three principal etiologies that may lead to this situation are chronic kidney disease (CKD), malabsorption syndromes, or chronic inadequate exposure to sunlight.
With CKD there is loss of 1-alpha-hydroxylase in the kidney, which results in a decreased conversion of 25-hydroxyvitamin D to the active 1,25-dihydroxyvitamin D. The low level of 1,25-dihydroxyvitamin D, with or without hypocalcemia, is detected by receptors on the parathyroid glands and results in increased PTH secretion, which is commonly seen in late-stage CKD (stages 3 to 5D).
In conditions such as Crohn disease, celiac disease, chronic pancreatitis, or Whipple disease, or following gastric bypass surgery, there is fat malabsorption that contributes to reduced absorption of vitamin D and dietary calcium (worsened if there is inadequate intake), ultimately leading to hypocalcemia and an increase in PTH.[2][9]
For most people, exposure to sunlight is the major source of vitamin D.[2] However, concerns of increased skin cancers due to ultraviolet radiation resulted in successful campaigns to minimize sun exposure during the precise times vitamin D can be synthesized in the skin.[10] Inadequate vitamin D exposure is common among older patients, particularly those who always use sunblock, are housebound, institutionalized, or hospitalized.[11] This may result in vitamin D deficiency, which can cause hypocalcemia and a consequent rise in PTH secretion.
Other causes include:
Diminished dietary intake of calcium
Increased calcium loss or increased metabolic requirement:
Bone growth in growing years
Pregnancy and breastfeeding
Bisphosphonate treatment
Idiopathic hypercalciuria
Loop diuretics
Rhabdomyolysis
Sepsis
Diminished PTH effect:
CKD
Pseudohypoparathyroidism (G-protein deficiency)
Pathophysiology
The metabolism of vitamin D is central to understanding the pathophysiology of the main causes of SHPT. There are two main forms of vitamin D: cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2).
[Figure caption and citation for the preceding image starts]: Vitamin D metabolismCreated by Dr Syazrah Salam; used with permission [Citation ends].
Cholecalciferol (vitamin D3) is produced in the skin after sun exposure, and is found in a limited number of foods.[12][13] Exposure to sunlight is the major source of vitamin D for most children and adults.[2] Cholecalciferol occurs naturally in relatively few foods: salmon, tuna, mackerel, and fish-liver oils are the best sources, with smaller amounts found in beef liver, cheese, and egg yolk. Ergocalciferol (vitamin D2) has a different side chain from cholecalciferol. It is also found naturally in small quantities in some mushrooms.[13] Both vitamin D2 and D3 are used to fortify milk, bread, and multivitamins in the US. In Europe, vitamin D3 is almost exclusively used for multivitamins and food fortification.
Once vitamin D (D2 or D3) is made in the skin or ingested in the diet, it undergoes two hydroxylations, the first in the liver to form 25-hydroxyvitamin D. This compound then enters the circulation bound to vitamin D-binding protein (DBP) and travels to the kidney where the megalin receptor translocates the DBP-25-hydroxyvitamin D complex into the renal tubule. Here, the enzyme 25-hydroxyvitamin D-1-alpha-hydroxylase (CYP27B) introduces a hydroxyl function to form 1,25-dihydroxyvitamin D.[2]
Fibroblast growth factor-23 (FGF-23) is an important regulator of vitamin D metabolism. FGF-23 inhibits 1-alpha-hydroxylase activity in the kidneys and increases 24-hydroxylase activity, which removes cholecalciferol and 1,25-dihydroxyvitamin D from the circulation. The overall effect is a reduction in the level of 1,25-dihydroxyvitamin D.[14]
In the intestines 1,25-dihydroxyvitamin D induces expression of an epithelial calcium channel, calcium-binding protein (calbindin), and a number of other proteins that help increase transport of calcium from the diet into the circulation.[2] 1,25-dihydroxyvitamin D also interacts with nuclear vitamin D receptor in the osteoblast to stimulate the expression of receptor activator of nuclear factor kappa-Β ligand (RANKL), which leads to osteoclast maturation and, thus, bone resorption. In this way 1,25-dihydroxyvitamin D helps maintain calcium homeostasis by increasing the efficiency of intestinal calcium absorption and mobilizing calcium stores from the skeleton.
Any deficiency in vitamin D causes a decrease in the efficiency of intestinal absorption of dietary calcium (and phosphorus).[15] This results in a transient lowering of the ionized calcium, which promptly triggers increased production and secretion of PTH.[9] PTH acts to increase ionized calcium in the blood by interacting with its membrane receptor on mature osteoblasts, which induces expression of RANKL. The RANKL protein is recognized by a protein called receptor activator of nuclear factor kappa-Β (RANK) present on the plasma membrane of preosteoclasts, and the RANKL-RANK interaction results in increased production and maturation of osteoclasts.[2] PTH also decreases the gene expression of osteoprotegerin (a decoy receptor for RANKL) in osteoblasts, which further enhances osteoclastogenesis. The osteoclasts release hydrochloric acid and collagenases to destroy bone, resulting in the mobilization of the calcium stores out of the skeleton. This vitamin D deficiency-induced SHPT results in the wasting of the skeleton, which can precipitate and exacerbate osteoporosis.[2]
Another effect of vitamin D deficiency and SHPT is loss of phosphorus in the urine and a lowering of serum phosphorus levels.[15] The inadequate calcium-phosphorus product that results causes the bone matrix laid down by osteoblasts to be abnormally mineralized. In children, the weight of the body causes the abnormally mineralized skeleton to develop classic rachitic deformities such as bowed legs or knocked knees. However, in adults there is generally sufficient skeletal mineralization to prevent skeletal deformities, although in a vitamin D-deficient state, newly laid-down osteoid cannot be properly mineralized, leading to osteomalacia.[15] This is associated with throbbing bone pain often misdiagnosed as fibromyalgia, myositis, or chronic fatigue syndrome. A possible explanation of the pain in these patients is that the poorly mineralized osteoid becomes hydrated and cannot provide sufficient support for the sensory fibers in the periosteum.[16] The pain commonly affects the pelvis, hips, legs, lower back, and ribs.
Chronic kidney disease-mineral bone disorder
Chronic kidney disease-mineral bone disorder (CKD-MBD) is defined as a systemic disorder of bone and mineral metabolism due to CKD manifested by either one or a combination of the following:[1]
Abnormalities of calcium, phosphorus, PTH, or vitamin D metabolism
Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
Vascular or other soft-tissue calcification.
The pathophysiology of CKD-MBD is complex. Principally there is a gradual decline in overall renal phosphate excretion, although paradoxically excretion per individual nephron rises sharply, which maintains serum phosphate in the normal range until CKD stage 4. There are also increased levels of PTH and FGF-23, two phosphaturic hormones that seem to accelerate the development of vascular disease particularly in the context of CKD.[17] Evidence for this comes from the Framingham Offspring study, which demonstrated that hyperphosphatemia was associated with increased risk of cardiovascular disease even in subjects free of CKD, and the Third National Health and Nutrition Examination Survey (NHANES).[18][19] Although FGF-23 is implicated in the pathophysiology of CKD-MBD, its use in clinical practice as a biomarker of disease progression is yet to be tested prospectively. There are also unresolved questions pertaining to assays, and the ability to make a sustained major reduction in serum FGF-23 concentrations remains elusive.
[Figure caption and citation for the preceding image starts]: Pathophysiology of secondary hyperparathyroidism in CKD-MBDCreated by BMJ Knowledge Centre from original flowchart by Dr Syazrah Salam; used with permission [Citation ends].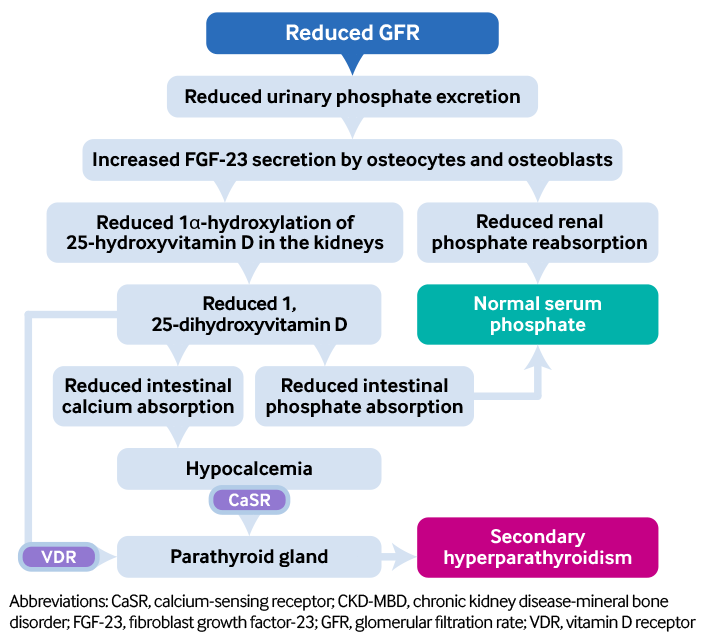
There is a spectrum of histomorphometric abnormalities that can be seen in patients with CKD, and to date inadequate clinical data and the heterogeneous nature of the condition have made it difficult to create a reliable and accurate classification system based on serum biochemical abnormalities alone to aid its diagnosis and management.[20] Indeed, dysfunctional bone mineral homeostasis and soft-tissue calcification are not exclusive to CKD and are multifactorial processes that may have more than one underlying etiology. For example, bone fragility, increased risk of fracture, and vascular calcification with atherosclerotic disease are associated with normal aging processes, and may occur in patients with normal or only mildly reduced renal function, only to exist concomitantly with CKD-MBD when renal function has deteriorated. Traditionally, a bone biopsy might have been conducted if there were biochemical inconsistencies, inexplicable bone pain, or fractures that would not support the diagnosis of CKD-MBD.[1] However, bone biopsy is very rarely undertaken in current clinical practice owing to its invasive nature.
Use of this content is subject to our disclaimer