Approach
Ataxia is characterized by clumsy and uncoordinated movement of the limbs, trunk, and cranial muscles, resulting from pathology in the cerebellum and its connections, or in the proprioceptive sensory pathways.
Cerebellar ataxia produces a characteristic set of signs, elicited from the history and exam:[108][109]
Gait is unsteady with a tendency to falls
Hand coordination is impaired
Dysarthria or dysphagia may be present
Ocular symptoms, related to abnormal control of eye movements, may occur.
A more detailed neurologic exam shows:
Dysmetria of limbs (inaccuracy of targeted movements) as shown by abnormal finger-chase test using patient fingers or toes
A kinetic tremor of limbs and an uncontrolled oscillation of limbs during relatively slow but targeted movements may be present
Early stance and gait problems, including the inability to do a tandem stance or stand with feet together; stance becomes broad-based and displays increased sway of the body. Tandem gait becomes impaired and, later, regular gait can be frankly ataxic with a broad-based and lurching quality
Eye movements that show gaze-evoked or other types of nystagmus, abnormal pursuit of visually presented objects (jerky appearance due to intrusion of saccades into pursuit), and inaccurate saccades when the person is asked to move the eyes quickly toward a target (hypometric or hypermetric saccades)
A scanning type of dysarthria.
Patients with ataxia as a major feature often exhibit additional noncerebellar neurologic signs, either because the lesion causing the ataxia extends beyond the cerebellum into neighboring structures, or because the degenerative condition involves structures beyond the cerebellum.
Sensory ataxia is characterized by lower-limb and upper-limb incoordination associated with lack of proprioception. Clinical signs include impaired vibration sense, as well as impaired position and kinesthetic sense. Deep tendon reflexes are often absent, but eye movements and speech are not affected. In some patients, sensory and cerebellar features can coexist.
Approach to differential diagnosis
Given the numerous disorders, as well as the extreme variability of presentation of ataxic diseases, it is useful to consider patients in some broad groups before attempting a specific diagnostic approach.
The broad groups of patients are as follows:
Patients with acute ataxia
Patients with subacute or chronic ataxia, with definitive structural abnormalities on imaging, with or without a positive family history
Patients with subacute or chronic ataxia in whom imaging shows atrophy of the cerebellum and associated structures (or, rarely, no abnormalities at all), with or without a family history.
The following factors are important modifiers of the diagnostic approach:
Age at onset
Tempo of disease
Presence or absence of a family history of the same disease
Whether ataxia is cerebellar or sensory in nature
Presence of nonataxic neurologic signs
Presence of certain systemic features
Presence or absence of overt structural abnormality by computed tomography (CT) or magnetic resonance imaging (MRI)
Acute ataxia
Patients present with acute onset of balance problems, with or without additional symptoms.
Age at onset is an important determinant of cause. Ataxia of acute onset in a child is most likely from acute cerebellitis or may be drug-induced (e.g., anticonvulsants). In adults, the most frequent causes are ischemic or hemorrhagic stroke in the cerebellum or brain stem, intoxications (such as therapeutic drugs, alcohol, and misuse of drugs), and Wernicke-Korsakoff syndrome. Demyelinating lesions such as multiple sclerosis can also have a rapid onset, and, if they occur in the cerebellum or its connections, present with acute ataxia. The Miller-Fisher variant form of Guillain-Barre syndrome needs to be considered.
History
Age at onset is a very important determinant.
Other critical pieces of information are the evolution of symptoms (months or a few years versus very chronic), an imaging study of the brain (preferably MRI), and a family history of ataxia.
If the family history is positive, the pattern of inheritance (autosomal recessive, autosomal dominant, X-linked) should be determined if possible, since this will narrow the differential diagnosis.
History of alcohol dependence and nutritional deficit should be obtained, as well as a complete drug history, including any misuse of drugs.
In patients with imbalance alone, history of vertigo, tinnitus, and hearing problems may indicate a peripheral vestibular problem.
Explore symptoms that indicate increased intracranial pressure (e.g., headache, nausea, vomiting) and symptoms that may indicate problems in structures contiguous to the cerebellum, such as the brain stem. These include weakness or sensory problems in the limbs, and cranial nerve deficits. Obtundation can be prominent with brain stem involvement.
Rarely in children and young adults, presentation may herald an illness in which repeated episodes of ataxia occur. History of such episodes and similar problems in other family members can point to an episodic ataxia syndrome. Inborn metabolic errors may have episodes of such ataxia as well.
Physical exam
Ataxia is incoordination of intentional movement.
Look for oscillations of the head and trunk, or titubation.
Evaluate the speech for dysarthria and scanning speech. In scanning speech, the volume of the patient’s voice varies from low to high.
Evaluate eye movements for nystagmus, jerkiness on attempted smooth pursuit, and slowed saccades. Oculomotor palsy can be associated with some disorders.
Primary position nystagmus and lateralized past pointing may suggest an acute vestibulopathy, but distinction between vestibulopathy and a small brain stem infarct can be difficult.
Look for neck stiffness.
Include a fundoscopic exam.
Evaluate the gait by checking for broad-based stance and gait. Test tandem walking.
Inspect for limb dysmetria by testing finger-to-nose, and heel-to-shin, dysdiadochokinesia, and heel-tapping test.
Check for overshooting with the wrist-tapping test, in which the patient is unable to maintain postures against an unexpected displacement.
The Miller-Fisher variant form of Guillain-Barre syndrome needs to be considered if there is ataxia, oculomotor palsy, and areflexia, with or without additional weakness in the limbs, bulbar muscles, and trunk.
Investigations
Workup with imaging of the brain (CT scan, MRI with diffusion-weighted imaging sequence) is required to look for signal changes associated with stroke, demyelination, or cerebellitis.[110][111][112][113] CT imaging in children necessitates the use of dedicated pediatric protocols to minimize radiation exposure.[113] [Figure caption and citation for the preceding image starts]: Acute bilateral cerebellar infarct, as seen on diffusion-weighted imaging sequence magnetic resonance imageFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].
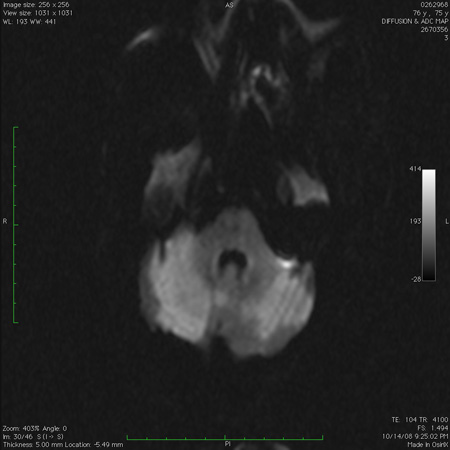 [Figure caption and citation for the preceding image starts]: Cerebellar infarct as seen on fluid-attenuated inversion recovery sequence magnetic resonance image: note secondary edema and effacement of the fourth ventricleFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cerebellar infarct as seen on fluid-attenuated inversion recovery sequence magnetic resonance image: note secondary edema and effacement of the fourth ventricleFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].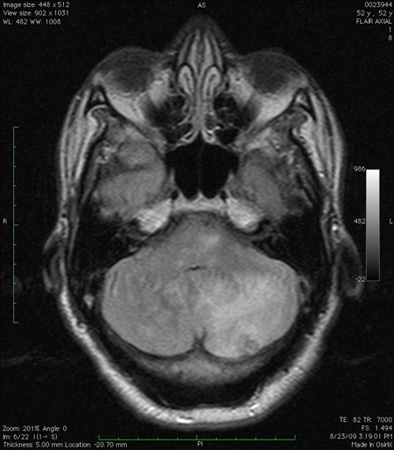 [Figure caption and citation for the preceding image starts]: Infarct in the dorsolateral medulla together with scattered infarcts in the cerebellum, as seen on diffusion-weighted imaging sequence magnetic resonance imageFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Infarct in the dorsolateral medulla together with scattered infarcts in the cerebellum, as seen on diffusion-weighted imaging sequence magnetic resonance imageFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].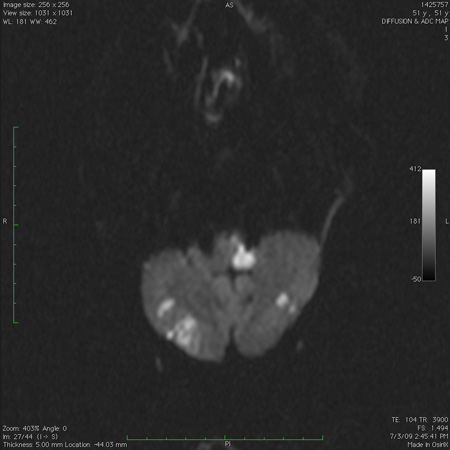 [Figure caption and citation for the preceding image starts]: Computed tomography scan of the brain showing a hemorrhage in the cerebellum with extension into the fourth ventricleFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Computed tomography scan of the brain showing a hemorrhage in the cerebellum with extension into the fourth ventricleFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].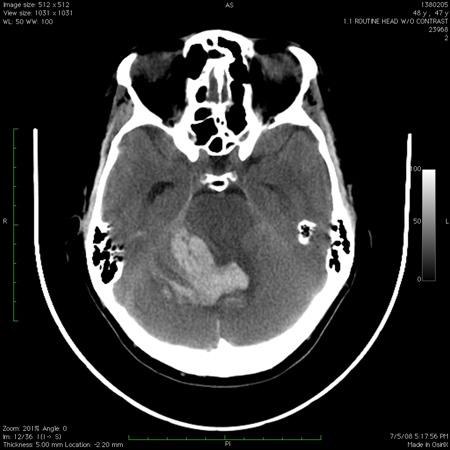
If brain imaging finds an ischemic infarct in the cerebellum or brain stem, studies to image the vascular system may need to be done acutely in selected cases; these include CT angiogram, magnetic resonance angiogram, and routine catheter angiography.[111]
Cerebrospinal fluid (CSF) exam for infectious/inflammatory parameters and IgG synthesis should be done if brain imaging findings:
fail to confirm an ischemic lesion or hemorrhage
are compatible with multiple sclerosis or cerebellitis.
Wernicke-Korsakoff syndrome is diagnosed primarily on a clinical basis and by a rapid response to vitamin B1 and other nutritional replacement.
Suspected Miller-Fisher syndrome patients need electrophysiologic examination of the neuromuscular system, and CSF exam, which may reveal high protein levels. Antibody to GQ1b is often elevated in Miller-Fisher syndrome, but results cannot be obtained in a rapid fashion.
Screening for substance misuse, including alcohol misuse, in patients with no structural lesions, and measurement of appropriate drug levels in those taking cerebellotoxic drugs, should be undertaken.
Subacute or chronic ataxia: definite structural abnormalities on imaging
Most of these patients are adults and have a clinical course that is relatively more rapid (measured in months or 1 or 2 years) than that seen in inherited forms of ataxia or sporadic/idiopathic ataxia. Also, family history of a similar disorder is usually negative. Such problems include vascular lesions (residuals of previous ischemic/hemorrhagic stroke, vascular malformations), demyelinating disease (multiple sclerosis), neoplastic lesions (primary and metastatic tumors), and compressive lesions of a non-neoplastic nature (i.e., craniovertebral junction anomalies).
Patients may have nervous system signs, often of a "contiguous" nature (signs of lesions in adjacent structures such as brain stem and upper cervical cord), of raised pressure, or signs in remote areas of the nervous system such as the optic nerve, hemispheres, and lower spinal cord (the latter indicates a demyelinating disease).
In any patient presenting with chronic/subacute ataxia, MRI of the brain with and without contrast should be done.[112] Most of the entities in this subgroup can be definitively diagnosed based on clinical history, the nature of clinical signs, and the imaging abnormalities. The only situations where such patients may have similar problems in the family are some rare familial tumor or malformation syndromes such as von Hippel-Lindau syndrome.[Figure caption and citation for the preceding image starts]: Large mass lesion in the cerebellum with pressure effects, as seen on MRIFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].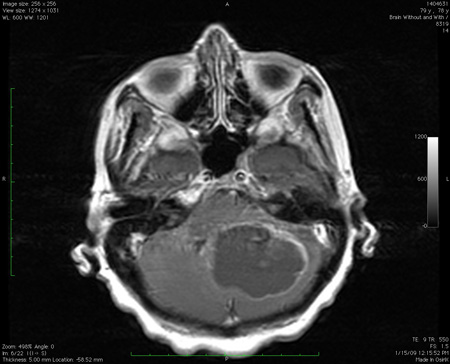
Subacute or chronic ataxia: cerebellar, brain stem, and spinal cord atrophy in varying combinations, or no imaging abnormality: negative family history
Childhood onset
Even when there is no family history, these children are likely to have a monogenic disease, since the various acquired causes are uncommon in children. Thus, the focus in this group is on uncovering a genetic etiology, either by DNA analysis or by finding an inborn metabolic error by biochemical testing, or both. Skin biopsy and fibroblast culture is required if Niemann-Pick disease type C is suspected.
Most of these disorders are autosomal-recessive entities. Other types of genetic diseases that may present in this group are those that have an X-linked or mitochondrial inheritance. Rarely, an autosomal-dominant ataxia can present in childhood before the affected parent becomes symptomatic; this is especially likely with SCA7 and dentatorubral-pallido-luysian atrophy (DRPLA). The details of the workup of these children are presented in the section below on childhood ataxia with a positive family history.
One form of acquired ataxia in children is the myoclonus-opsoclonus syndrome, which is characterized by the rapid or subacute onset of ataxia, myoclonic jerks, and chaotic eye movements of an involuntary nature. This is believed to be autoimmune in origin and may be triggered by an underlying malignancy, usually a neuroblastoma. Imaging studies of the brain and abdomen and CSF exam may be helpful.
Adult onset
Acquired causes of ataxia predominate in this group and should be excluded as the first priority.
This is the average adult "singleton" patient with ataxia in whom family history is negative for a similar disease. To be sure of a negative family history, it is useful to construct a pedigree with at least 3 generations and enquire into possible similar disease in as many relatives as possible. Parental age at death may be important because a negative family history may be due to the early death of an affected parent.
History should explore alcohol misuse, nutritional status (e.g., weight loss, chronic gastrointestinal disease), endocrine symptoms, history of significant hypoxia or heat stroke, medications being taken (including over-the-counter drugs), drug misuse, and the presence of organ-specific autoimmune disease such as diabetes or hypothyroidism. History or risk factors for HIV infection should be obtained.
Physical exam should determine whether the ataxia is cerebellar or sensory in nature. Sensory ataxias are less common and have a very differential diagnosis. Ataxia in this group of patients (i.e., with an atrophic process on imaging) tends to be symmetric on both sides of the body. Ataxia in patients with overt lesions detected by imaging can often be asymmetric.
Investigations of value include an MRI study of the brain, which excludes structural lesions and documents cerebellar atrophy with or without brain stem or upper spinal cord atrophy. Thyroid studies, vitamin levels (i.e., B1, B6, B12), methylmalonic acid and homocysteine levels (which indicate B12 status), drug levels (e.g., phenytoin, carbamazepine, phenobarbital as indicated), antibodies to glutamic acid decarboxylase, gliadin, and endomysium (tissue transglutaminase), parietal cell antibodies, paraneoplastic antibodies, HIV serology, polymerase chain reaction for Whipple disease, monoclonal spike studies (such as serum immunoelectrophoresis), anti-MAG antibodies, intestinal biopsy, and peripheral nerve electrophysiology may all contribute to the diagnosis. Specific tests should be selected based on clinical information.[Figure caption and citation for the preceding image starts]: MRI of brain showing early cerebellar and brain stem atrophy in SCA 1From the collection of Dr S. H. Subramony; used with permission [Citation ends].
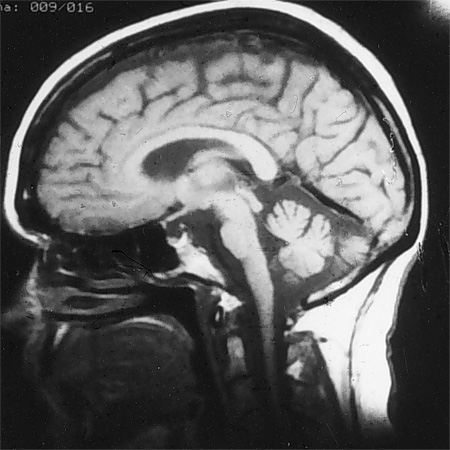
If the clinical picture is suspicious, a search should be performed for a visceral cancer such as that of lung, ovary, or breast, including full physical exam and scans.
While nongenetic diseases dominate in this group, in people in whom one of the above diagnoses cannot be established, an autosomal-recessive, X-linked, or mitochondrial mutation needs to be looked for. In selected cases, autosomal-dominant mutation analysis may also be done, if the clinical picture is worrisome for one of these and there is concern that the family history is not reliable. The common DNA abnormalities that have been seen in series of such patients include Friedreich ataxia, fragile-X tremor-ataxia syndrome, SCA6, SCA8, and mitochondrial DNA mutations.
In addition, some of the "inborn errors of metabolism" may rarely present in this age group. Laboratory studies that may point to some of these disorders include serum CK, alpha-fetoprotein, lactate and pyruvate levels, very long-chain fatty acid levels, phytanic acid levels, plasma chitotriosidase, and ammonia levels. The yield for individual analyses is small, however.
Subacute or chronic ataxia: cerebellar, brain stem, and spinal cord atrophy in varying combinations or no imaging abnormality: positive family history
Childhood onset
Most of these are autosomal-recessive diseases, but X-linked, mitochondrial, and autosomal-dominant ataxias may also present in childhood. While autosomal-recessive inheritance is indicated by affected siblings and parental consanguinity, many of these children present as singleton patients. Many autosomal-recessive ataxias may occasionally present for the first time in adult life and need to be considered in adults as well.
Rarely, autosomal-dominant ataxias can present in childhood before an affected parent becomes symptomatic (e.g., SCA7 and DRPLA).
Age at onset should be determined. Some entities, such as ataxia with oculomotor apraxia 1 (AOA1) and ataxia telangiectasia (AT), begin in the first decade, while the more common Friedreich ataxia and ataxia with oculomotor apraxia 2 (AOA2) typically have their onset in the second decade.
Many of these cases, including most children with Friedreich ataxia, have absent tendon reflexes, reflecting coexistent peripheral nerve disease. Rarer diseases can have associated spasticity and brisk tendon reflexes. Eye movement apraxia is seen (but not universally) in AT, AOA1, and AOA2. Associated choreic movements are also seen in AT, AOA1, and AOA2, but not in Friedreich ataxia. Head tremor and retinopathy can be seen in ataxia with vitamin E deficiency (AVED), and retinopathy in mitochondrial DNA abnormalities. Pes cavus may be seen in Friedreich ataxia. The patient should be examined for systemic abnormalities.[Figure caption and citation for the preceding image starts]: Pes cavus deformity in Friedreich ataxiaFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].
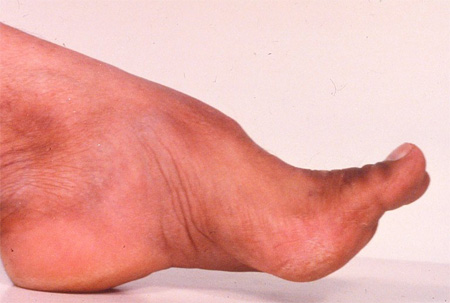
Unlike autosomal-dominant ataxias, autosomal-recessive ataxias often show affected non-central nervous system tissues. Examples include cardiomyopathy and diabetes in Friedreich ataxia, conjunctival and cutaneous telangiectasia in AT, lymphoreticular malignancy in AT, elevated serum alpha-fetoprotein in AT and AOA2, low serum albumin in AOA1, Kayser-Fleischer rings and liver disease in Wilson disease, and tendon xanthomas and cataracts in cerebrotendinous xanthomatosis.[Figure caption and citation for the preceding image starts]: Early conjunctival telangiectasia in a patient with ataxia telangiectasiaFrom the collection of Dr S. H. Subramony; used with permission [Citation ends].
 [Figure caption and citation for the preceding image starts]: Tendon xanthoma seen in cerebrotendinous xanthomatosisFrom the collection of Dr S. H. Subramony, with thanks to Dr Uday Muthane; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Tendon xanthoma seen in cerebrotendinous xanthomatosisFrom the collection of Dr S. H. Subramony, with thanks to Dr Uday Muthane; used with permission [Citation ends].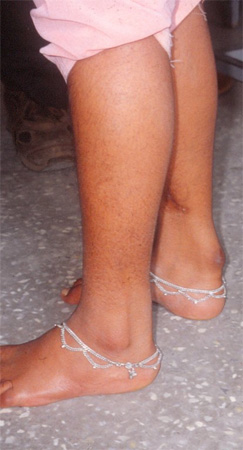
In these children, MRI of the brain and upper spinal cord should be done first. High cord atrophy is seen in Friedreich ataxia. Cerebellar atrophy is seen in AT, AOA1, and AOA2. Peripheral nerve electrophysiology may point to prominent nerve involvement, seen in Friedreich ataxia and to a lesser degree in AVED and the AOAs.
The most common autosomal-recessive mutation is the FA GAA expansion (confined to Indo-European populations), then AOA2. AT can often be clinically diagnosed and then confirmed either by targeted mutation analysis or by protein truncation tests. Radiosensitivity of cultured fibroblasts is another test that can be employed.
Since AVED can be treatable, serum tocopherol levels should be obtained in any child or adult with chronic ataxia of otherwise undetermined cause.
Other autosomal-recessive mutations that can be obtained include those causing AOA1, mitochondrial-recessive ataxia syndrome, autosomal-recessive spastic ataxia of Charlevoix-Saguenay, and AVED.
A mitochondrial DNA abnormality should be considered next. Exclude an autosomal-dominant ataxia by careful family history, examination of the parents, and mutation analysis (common considerations SCA7, SCA2, and DRPLA, the latter being more common in Japan) if necessary.
Since genetic disorders with "inborn errors of metabolism" often present in this age group, such diseases need to be excluded by metabolic or DNA studies. Laboratory studies include serum and urine amino acids, urine organic acids, serum ammonia, tests for lysosomal storage diseases, and serum cholestanol. Other useful tests may include magnetic resonance spectroscopy to detect vanishing white matter disease, serum ceruloplasmin and copper levels for Wilson disease, and phytanic acid levels. These are best done in collaboration with a metabolic disease expert.
Adult onset
Most commonly, onset in this age group is associated with a family history consistent with autosomal-dominant inheritance (SCAs).
In this scenario, an imaging study may be done to exclude a coincidental structural lesion, but this may not be needed if the clinical picture is entirely consistent and the clinician is knowledgeable about other family members.
If the mutation in the family is already established, another family member with the typical clinical signs as having the same disease can be diagnosed by clinical criteria alone, or by obtaining only the mutation already known in the family. If the family mutation is unknown, the clinical picture and prevalence pattern of different SCAs can be useful in determining the most likely genotype, although all experts agree that one cannot make a genotypic diagnosis based on the phenotype alone.
The most common SCA worldwide is SCA3, also known as Machado-Joseph disease (MJD). Certain parts of the world, such as Portugal (especially the Azores) and some previously Portuguese-occupied territories such as Brazil, have an even higher prevalence of MJD. This is followed by SCA2, SCA6, and SCA1, probably in that order for most areas of the world. All of these, together with SCA7 (in which there is associated retinopathy and visual loss), are caused by unstable CAG expansions in their respective genes and probably should be the first line of mutation analysis in a patient with SCA.
SCA1, SCA2, and SCA3 have a wide range of age of onset, but the mean is in the third decade. The disease course is steadily progressive, with death commonly in the fifth decade. In contrast, SCA6 generally has a later onset in the fourth decade, and a disease course often compatible with a normal lifespan. SCA7 and DRPLA have both childhood and adult onset; the disease course is more aggressive with early onset. Visual loss in SCA7 is not universal, but tends to occur with earlier onset.
A good family history with details of the phenotype in other family members may indicate the genotype. Once the CAG expansions are excluded, mutation analysis might be considered for the CTG expansion of SCA 8, other nucleotide expansion disorders such as SCA12, SCA10, and SCA17, and those in which other mutational mechanisms have been uncovered (e.g., SCA13, SCA14).
Still other SCAs have had mutations uncovered recently, but these are often available for testing only in research laboratories. Interestingly, one study using a novel technique of next-generation exome sequencing in patients with adult onset and sporadic presentations demonstrated that this is a high-yield test, giving a definitive diagnosis in more than one fifth of patients, and suggesting a potential diagnosis in more than one third. The study posits that, in future, clinical exome sequencing may be considered as part of the routine evaluation of all patients presenting with chronic progressive cerebellar ataxia.[114]
Adult-onset ataxia with clear autosomal-recessive inheritance is less common, but will need workup as for children with this type of ataxia.
Finally, adult-onset (usually >50 years) ataxia with an atrophic process on imaging studies, with no family history, no mutation established, and all known acquired causes ruled out, is labeled as sporadic or idiopathic ataxia. In this group, detection of dysautonomia by detailed autonomic function tests and anal sphincter denervation by electromyogram may allow a diagnosis of probable multiple system atrophy.
Disorders resembling ataxia
Many types of gait problems can superficially resemble ataxia. Examples include gait abnormalities from bilateral frontal lobe lesions (i.e., frontal ataxia or Brun ataxia) and, occasionally, dystonic gait. Gait disorders from muscle disease, neuropathies, and spinal cord problems can usually be easily differentiated by clinical exam.
Gait difficulties from lesions of the vestibular apparatus also need to be differentiated from cerebellar ataxia. The coordination problem in the limbs is in the nature of "past pointing" rather than true dysmetria, and eye movement problems are often confined to nystagmus in the primary position. Dysarthria is not a feature of vestibular disease, but nausea and oscillopsia can be a feature of vestibular lesions.
The presence of speech and oculomotor signs related to cerebellar pathology can clearly differentiate cerebellar ataxia from other types of incoordination.
Use of this content is subject to our disclaimer