Aetiology
The most common cause of achlorhydria is chronic atrophic gastritis. Gastric atrophy may be defined as the loss of glands (oxyntic glands) and/or parietal cells in the fundus and corpus (body) of the stomach. The glands are typically replaced by connective tissue (non-metaplastic atrophy) or glandular structures inappropriate for the location (metaplastic atrophy).[32]
Causes of gastric atrophy are Helicobacter pylori infection, primary autoimmune gastritis, and a combination of H pylori and autoimmune gastritis.[33] Different gastric microbiome profiles have been reported in patients with H pylori-induced gastritis and those with autoimmune gastritis.[34]
Gastric atrophy, with or without autoimmune gastritis, is thought to be, for the most part, initiated by infection with H pylori.[35][36][37][38] It is proposed that H pylori-induced inflammation results in breakdown of tolerance for self antigens (e.g., hydrogen-potassium-stimulated adenosine triphosphatase [H+/K+ ATPase]) in genetically susceptible individuals, or that antibodies are acquired due to molecular mimicry between H pylori lipopolysaccharide and H+/K+ ATPase, both of which contain Lewis epitopes. The pathology initially shows a chronic inflammatory, mainly lymphocytic, infiltrate of the oxyntic mucosa, accompanied by loss of parietal and chief cells. Later, marked atrophy of the oxyntic glands develops with only scattered lymphoid aggregates.
Other causes of achlorhydria include antrectomy with vagotomy for peptic ulcer disease and subtotal gastrectomy for cancer. It should be noted that proton-pump inhibitors do not completely inhibit acid secretion over 24 hours due to their relatively short plasma half-life (1 to 2 hours), their limited residence time in the systemic circulation, and the regeneration of approximately 25% of pumps each day.[39][40][41]
Gastric intestinal metaplasia
Gastric atrophy is invariably associated with some degree of gastric intestinal metaplasia (IM) and hypergastrinaemia. Gastric IM is a pre-malignant condition with a small increased annual risk of adenocarcinoma (0.1% to 0.2%), which is characterised by a change in the gastric mucosa to a small-intestinal phenotype.[23][42] H pylori infection is the strongest and most consistent risk factor for gastric IM (threefold-eightfold increased risk).[43][44]
Observational data suggest an association between male sex, older age, non-white ethnicity, current smoking status, and increased presence of gastric IM.[45] Among patients with gastric IM, male patients and those with incomplete gastric IM may be at greatest risk of developing dysplasia or early gastric cancer.[46]
[Figure caption and citation for the preceding image starts]: Model illustrating physiological regulation of gastric acid secretion by gastrin, histamine, somatostatin (SST), and luminal acid. Gastrin, released from antral G cells, is the main hormonal stimulant of acid secretion during meal ingestion. Gastrin acts directly on the acid-secreting parietal cells and, more importantly, indirectly by stimulating histamine secretion from enterochromaffin-like (ECL) cells. Histamine diffuses to adjacent parietal cells, where it binds to histamine H2 receptors coupled to stimulation of acid secretion. In the interdigestive phase, somatostatin (SST), released from antral D cells in response to luminal acid, tonically inhibits gastrin secretion from G cells, thereby maintaining acid secretion at an economically low level.From the collection of Professor Mitchell L. Schubert, with the acknowledgement of Mary Beatty-Brooks (medical illustrator) [Citation ends].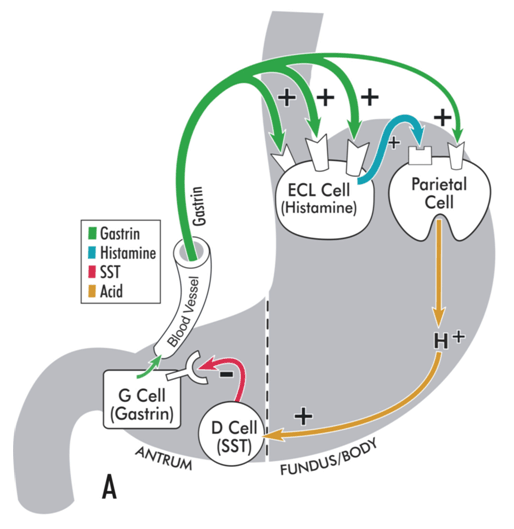 [Figure caption and citation for the preceding image starts]: Model illustrating pathophysiology of achlorhydria and the development of gastric carcinoid tumours. With achlorhydria, the stimulatory effect of luminal acid on SST is lost. Consequently, SST secretion is decreased and its inhibitory restraint on gastrin secretion attenuated (disinhibition), resulting in hypergastrinaemia. Gastrin is not only a secretagogue but also a trophic hormone that induces growth of the oxyntic mucosa. If hypergastrinaemia is sustained for days, ECL cells will hypertrophy; if sustained for weeks to months, ECL cells become hyperplastic, dysplastic, and, in some patients, become carcinoid tumours.From the collection of Professor Mitchell L. Schubert, with the acknowledgement of Mary Beatty-Brooks (medical illustrator) [Citation ends].
[Figure caption and citation for the preceding image starts]: Model illustrating pathophysiology of achlorhydria and the development of gastric carcinoid tumours. With achlorhydria, the stimulatory effect of luminal acid on SST is lost. Consequently, SST secretion is decreased and its inhibitory restraint on gastrin secretion attenuated (disinhibition), resulting in hypergastrinaemia. Gastrin is not only a secretagogue but also a trophic hormone that induces growth of the oxyntic mucosa. If hypergastrinaemia is sustained for days, ECL cells will hypertrophy; if sustained for weeks to months, ECL cells become hyperplastic, dysplastic, and, in some patients, become carcinoid tumours.From the collection of Professor Mitchell L. Schubert, with the acknowledgement of Mary Beatty-Brooks (medical illustrator) [Citation ends]. [Figure caption and citation for the preceding image starts]: Classification of gastric carcinoid tumoursHou W, Schubert, ML. Treatment of gastric carcinoids. Curr Treat Options Gastroenterol. 2007;10:123-133. Used with kind permission from Current Medicine Group LLC [Citation ends].
[Figure caption and citation for the preceding image starts]: Classification of gastric carcinoid tumoursHou W, Schubert, ML. Treatment of gastric carcinoids. Curr Treat Options Gastroenterol. 2007;10:123-133. Used with kind permission from Current Medicine Group LLC [Citation ends].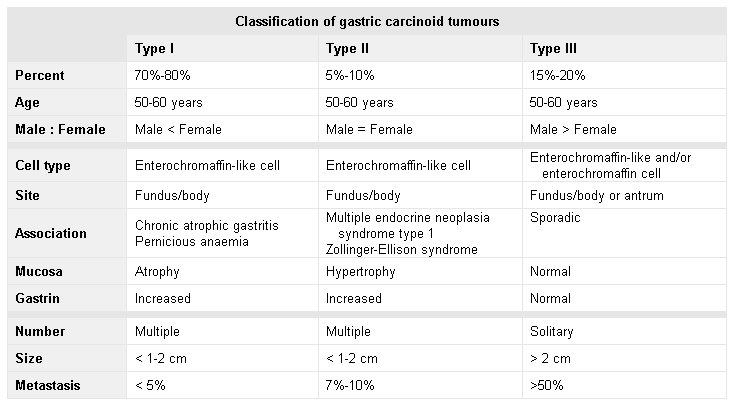
Pathophysiology
Helicobacter pylori colonises half the world's population, and most chronically infected patients manifest a pangastritis and produce less than normal amounts of acid.[47]
Reduced acid secretion, initially, may be attributable to H pylori (e.g., functional inhibition of the gastric proton pump [H+/K+ ATPase]) or to cytokines released from the inflammatory infiltrate (e.g., direct inhibition of parietal cell secretion by interleukin-1beta and tumour necrosis factor-alpha).[1][48][49][50][51]
With time, atrophy of oxyntic glands with loss of parietal cells may occur, resulting in irreversible achlorhydria. There is mounting evidence that H pylori may play a role in the development of autoimmune gastritis, and its resultant atrophic gastritis, in susceptible individuals. It is postulated that antibodies directed against H+/K+ ATPase occur in a subset of patients infected with H pylori as a result of molecular mimicry between the organism's lipopolysaccharide and the proton pump, both of which express Lewis x and y blood group antigens.[1][37][38]
Autoimmune gastritis is also associated with other diseases thought to be of immunological origin, such as: autoimmune thyroid disease (Graves' disease and Hashimoto’s thyroiditis [also termed autoimmune thyroiditis and chronic lymphocytic thyroiditis]); type 1 diabetes mellitus; idiopathic adrenocortical insufficiency; vitiligo; hypoparathyroidism; and primary biliary cholangitis.[52][53][54][55][56][57] In autoimmune thyroid disease, serological oxyntic atrophy is present in about 25% of cases.[58] In primary biliary cholangitis, anti-parietal cell antibodies and anti-intrinsic factor antibodies are present in 32% and 12% of cases, respectively.[52]
Surgical interventions such as antrectomy with vagotomy may induce achlorhydria. The principal stimulants of acid secretion are gastrin (released from G cells of gastric antrum), histamine (released from enterochromaffin-like [ECL] cells of gastric body/fundus), and acetylcholine ([ACh] released from postganglionic neurons located within the wall of the stomach).[1] Gastrin also functions as the main hormone responsible for growth of the oxyntic mucosa. Antrectomy removes gastrin, the main hormonal stimulant of acid secretion during ingestion of a meal. Vagotomy removes the effect of centrally acting stimulants of acid secretion such as the thought, sight, smell, and taste of food. Bile refluxed into postsurgical stomach remnants can induce chronic inflammation and atrophy. [Figure caption and citation for the preceding image starts]: Model illustrating physiological regulation of gastric acid secretion by gastrin, histamine, somatostatin (SST), and luminal acid. Gastrin, released from antral G cells, is the main hormonal stimulant of acid secretion during meal ingestion. Gastrin acts directly on the acid-secreting parietal cells and, more importantly, indirectly by stimulating histamine secretion from enterochromaffin-like (ECL) cells. Histamine diffuses to adjacent parietal cells, where it binds to histamine H2 receptors coupled to stimulation of acid secretion. In the interdigestive phase, somatostatin (SST), released from antral D cells in response to luminal acid, tonically inhibits gastrin secretion from G cells, thereby maintaining acid secretion at an economically low level.From the collection of Professor Mitchell L. Schubert, with the acknowledgement of Mary Beatty-Brooks (medical illustrator) [Citation ends].[Figure caption and citation for the preceding image starts]: Model illustrating pathophysiology of achlorhydria and the development of gastric carcinoid tumours. With achlorhydria, the stimulatory effect of luminal acid on SST is lost. Consequently, SST secretion is decreased and its inhibitory restraint on gastrin secretion attenuated (disinhibition), resulting in hypergastrinaemia. Gastrin is not only a secretagogue but also a trophic hormone that induces growth of the oxyntic mucosa. If hypergastrinaemia is sustained for days, ECL cells will hypertrophy; if sustained for weeks to months, ECL cells become hyperplastic, dysplastic, and, in some patients, become carcinoid tumours.From the collection of Professor Mitchell L. Schubert, with the acknowledgement of Mary Beatty-Brooks (medical illustrator) [Citation ends].
Classification
Classification of gastric atrophy
Standard classification does not exist for achlorhydria, but classification systems exist for the classification of gastritis, including chronic active gastritis.
Gastric corpus atrophy is defined as the loss of gastric glandular cells, with and without its replacement with fibrosis, intestinal metaplasia (IM), or pseudopyloric/pyloric metaplasia. In IM, the normal epithelium is replaced by intestinal epithelium that contains goblet and/or Paneth cells. In pseudopyloric metaplasia, also termed spasmolytic polypeptide-expressing metaplasia (SPEM), oxyntic glands are replaced by antral-like glands.
Four staging systems are interchangeably used to classify gastric atrophy, based on the extension and severity of histological changes: updated Sydney system, operative link on gastritis assessment (OLGA), Baylor system, and operative link on intestinal metaplasia assessment (OLGIM).
Updated Sydney system[8]
Intended to identify and resolve issues with the Sydney system (that emphasised the importance of combining topographical, morphological, and aetiological information to generate reproducible and clinically useful diagnoses). The final classification was improved to highlight the distinction between the atrophic and non-atrophic stomach.
Operative link on gastritis assessment (OLGA)[5][9][10]
The OLGA system integrates atrophy score and atrophy topography in one system. The grading system has been validated in one long-term follow-up study of 7436 patients; higher stages at the time of enrolment carried a higher risk of gastric dysplasia on follow-up.[11]
Baylor system[12]
Non-invasive testing with serum concentrations of pepsinogens and gastrin are used as indirect measures of gastric oxyntic mucosal functional integrity.
Operative link on intestinal metaplasia assessment (OLGIM)[10][13]
Recognises IM in gastric mucosa in an easier and more consistent manner than the OLGA system.
Use of this content is subject to our disclaimer