Approach
A detailed history is of paramount importance, as epileptic seizures are often not witnessed by a physician. A description of the seizure onset is key (to help categorise as focal-onset or generalised-onset seizure), as well as additional information such as: the setting, any possible provoking factors, warning signs that the child or young person may be able to recount, and any postictal phenomena. An electroencephalogram (EEG) is used to help determine the specific epilepsy syndrome and identify features such as photosensitivity.[18][19] Standard laboratory tests such as fulll blood count and basic metabolic panel (blood sugar, calcium, magnesium) are not routinely indicated, but may be requested initially to exclude any underlying causes. Brain imaging may be indicated if the onset of the seizure has not been witnessed or if there is suggestion of a focal onset; however, in most cases of generalised epilepsies with a clear electroclinical syndromic diagnosis, brain imaging is not indicated. Genetic testing is recommended if certain epilepsy syndromes are suspected.
The ILAE Commission on Classification and Terminology has developed an online diagnostic manual of the epilepsies to assist clinicians to diagnose seizure type(s), diagnose epilepsy syndromes, and define the aetiology of the epilepsy. ILAE: EpilepsyDiagnosis.org Opens in new window
History
History can be elicited from the parents or any eyewitnesses. Family history of seizures or epilepsy, past medical history, birth history, and the child's developmental history should be noted.
It is important to go through the events in chronological order, exploring the circumstances of the seizure, what the child was doing at the time, possible triggers (e.g., light, noise, sleep deprivation, fatigue, emotional state), presence of aura or other warnings (e.g., déjà vu, dizziness, altered vision), and how the seizures started and what they looked like. Video recording an episode or capturing the event on a smartphone is very useful to the clinician. Alternatively, demonstrating some of the movements or asking any witnesses to mimic what they saw is usually extremely helpful in determining the type of seizure (i.e., myoclonic, atonic, tonic and/or clonic, or absence). Traditionally the following have been thought to signify an epileptic seizure: specific movements; tone; state of consciousness; presence of incontinence, tongue biting, or eye movements, as well as how the seizure ended and if there were any signs/symptoms (e.g., sleepiness, headache, amnesia, confusion) in the post-ictal state. However, these on their own do not equate to a person having had an epileptic seizure.
Seizure types
Characteristics of seizure types include:[2]
Absence: impairment (usually brief) of awareness, which may be associated with motor arrest or with stereotyped movements or automatisms. It is important to differentiate between the two types of absence seizure, as they occur in different epileptic syndromes:
Typical absence seizure: behavioural arrest or staring, lasting 5-10 seconds, interrupting otherwise normal activity; can be hyperventilation-induced
Atypical absence seizure: less distinct beginning and end; not usually precipitated by hyperventilation.
Myoclonic: brief, arrhythmic muscular jerking movements.
Clonic: rhythmic, muscular jerking movements with or without impaired consciousness.
Tonic: tonic extension or flexion of the extremities.
Tonic-clonic: tonic phase involves the patient falling unconscious, possibly falling to the ground, and extension or flexion of extremities, and may be preceded by aura; clonic phase consists of usually violent muscle contractions and shaking.
Atonic: brief loss of muscle tone causing what are known as 'drop attacks', where the patient falls to the ground.
Length of seizures may vary from one event to another in the same person, but length can also be a specific characteristic of a seizure. For example, absence seizures typically last for up to 20 seconds, and a longer duration is so exceptional that it would warrant questioning the diagnosis.[20] Most generalised tonic-clonic seizures last <5 minutes. Differentiating between these and generalised tonic-clonic seizures that last longer is not helpful diagnostically, but informs immediate management decisions.
Sometimes seizures can be induced by light. This is called photosensitivity and is a useful diagnostic characteristic. The following syndromes are known to be photosensitive: epilepsy with eyelid myoclonia, juvenile myoclonic epilepsy, epilepsy with generalised tonic-clonic seizures alone, Dravet syndrome, and progressive myoclonic epilepsies.[21][22]
Physical examination
After an unprovoked seizure, the possibility of an underlying disorder should be considered during the clinical assessment. The physician should look for neurocutaneous signs, as conditions with these signs can manifest with seizures, including tuberous sclerosis complex (i.e., depigmented macules, shagreen patches, periungual fibromas, adenoma sebaceum, and ash-leaf macules), neurofibromatosis type 1 (i.e., cafe au lait spots, axillary and inguinal freckling, and plexiform neurofibromas), and Sturge-Weber syndrome (i.e., port wine-coloured haemangiomas on the face or trunk).[12]
Full neurological examination is an important part of the evaluation. Blood pressure, head circumference measurement, and an ECG is required for any child who has had a generalised seizure. In the majority of patients with recurrent generalised seizures there are no abnormalities.
Electroencephalogram (EEG)
EEG is a standard investigation for diagnosis of epilepsy.[23] Epileptic discharges may be seen on the inter-ictal recording, and abnormal rhythms can be characteristic for a specific epilepsy syndrome.[18][19] About 5% of children without epilepsy have non-specific abnormalities on EEG, and up to 40% of children with epilepsy have normal inter-ictal EEGs. Therefore, EEG complements history, physical, and other evaluation techniques. A normal inter-ictal EEG does not exclude a diagnosis of epilepsy. Epileptiform activity in a combined wake and sleep recording increases the probability that the diagnosis of epilepsy is the correct one from 0.17 (for children without these abnormalities) to 0.95.[24]
Hyperventilation, photic stimulation, and sleep deprivation may be used to provoke seizures as part of the EEG investigation or to enhance EEG abnormalities. For childhood absence epilepsy, inducing a seizure in the clinic by asking the child to over-breathe for up to 3 minutes is a useful diagnostic tool, as it will result in an absence seizure in 90% of children with absence epilepsy.[20]
A video EEG may be useful. It is used to determine whether the clinically observed seizures are epileptiform in nature. It can also help to characterise the seizures better and therefore clarify the diagnosis. Some studies suggest that an EEG is useful in predicting the risk of seizure recurrence in children and adolescents.[25][26]
Electrocardiogram (ECG)
An ECG should be performed for all children presenting with a first seizure to exclude cardiac causes of clinical events, primarily long QT syndrome.[27][28] Other possible causes include Brugada syndrome and possible arrhythmias. A cardiologist should be consulted if there is doubt or if there is a family history of early unexplained death.
Neuroimaging
The combination of typical EEG findings that correlate with the clinical picture can often be diagnostic. However, in cases where the diagnosis is in doubt or a secondary cause is suspected, magnetic resonance imaging (MRI) is the definitive imaging technique in children, as it does not involve ionising radiation and the images give more detail than computed tomography (CT) scans.
CT has a role in acute investigation for children with a recent history of a seizure and in whom an intracerebral bleed needs to be ruled out. It is also used when there are concerns about a space-occupying lesion and an MRI is not available.[23][29]
Functional MRI may be used for pre-surgical mapping.[30]
Laboratory investigations
These are not routinely indicated for patients with recurrent seizures. However, blood glucose level, basic metabolic panel, and a full blood count may be considered in children with suspected provoking factors such as hypoglycaemia, electrolyte abnormalities, metabolic disorders, or infection. Blood glucose measurement is an obligatory part of evaluation for any child with decreased consciousness level. It is recommended that these tests are ordered at least once for all patients with recurrent seizures.
Genetic testing
Genetic testing is recommended for children with suspected Dravet syndrome. It should also be considered for suspected developmental and epileptic encephalopathies (including suspected early infantile developmental and epileptic encephalopathy, infantile epileptic spasm syndrome, and Lennox-Gastaut syndrome), or if structural abnormalities suggest a genetic cause.[21][31][32][33]
Diagnosis of specific epilepsy syndromes
Patient history (including seizure types), EEG, physical examination, and complementary diagnostic information can help classify epilepsy into specific electroclinical epilepsy syndromes. Some syndromes will have a normal EEG, especially at initial presentation, so history and presentation are also vital. Identifying epilepsy syndromes provides guidance on management, outcomes, comorbidities, and potential additional evaluations. The patient's age may also guide diagnosis, since the incidence of specific syndromes peaks during particular age ranges.[1]
Onset in infancy (age 1 month to 2 years)[31]
Early infantile developmental and epileptic encephalopathy (EIDEE; includes Ohtahara syndrome):
Onset is usually by age 3 months (adjusted for prematurity).
Tonic and/or myoclonic seizures are typical, but other seizure types may occur.
EEG may show burst-suppression, multifocal spikes/spike-waves/sharp-waves with or without slowing, discontinuity, and/or diffuse slowing. The burst-suppression pattern comprises high-voltage bursts of mixed spikes, and sharp and slow waves lasting 1-5 seconds, alternating with periods of marked suppression lasting 3-10 seconds, and may be seen when the patient is awake or asleep. De-synchronisation is seen during tonic spasms.
Abnormal neurological behaviour or development may be present before onset of seizures (but this may be difficult to assess).
Causative pathogenic gene variants can be identified in over half of patients.
Infantile epileptic spasms syndrome (IESS):
Onset of epileptic spasms is usually between 1 and 24 months of age (peak 3-12 months), but may be later.
Characterised by flexor, extensor or mixed epileptic spasms, which often occur in clusters.
EEG: hypsarrhythmia (asynchronous asymmetrical spikes and slow waves) is usually, but not always, seen on inter-ictal recordings.
Development may or may not be normal before onset, but developmental slowing, arrest, or regression is seen after onset.
A sub-group with the triad of infantile spasms, developmental stagnation or regression, and hypsarrhythmia on EEG is defined as West syndrome.
Many pathogenic gene variants have been associated with IESS.
Myoclonic epilepsy in infancy (MEI):
Onset is usually between ages 4 months and 3 years (peak 6-18 months).
Characterised by myoclonic seizures that occur multiple times a day both during sleep and when awake. They may be triggered by noise, startle, touch, or (less commonly) photic stimulation. The seizures may be brief and singular, or occur in clusters.
EEG: background is normal, but generalised poly-spike or spike-and-slow-wave discharges are seen during myoclonic seizures.
Development is usually normal before seizure onset. A majority of patients show normal developmental outcome, but mild intellectual disability or behavioural problems may be seen. Moderate to severe intellectual disability is rare.
Dravet syndrome:
Onset by age 1 year, with characteristic prolonged, febrile and afebrile, focal clonic (usually hemiclonic), or generalised clonic seizures.
Other seizure types (e.g., myoclonic, atonic, tonic-clonic, and/or atypical absence seizures) may appear between the ages of 1 and 4 years.
EEG is initially normal. After age 2 years, slowing is typical, and inter-ictal discharges are often focal, multifocal, and generalised. Ictal recordings depend on seizure type.
Children typically have normal development before onset of seizures. Developmental plateau and other neurological abnormalities, such as ataxia, often present during the second year of life.
Frequently, there is a family history of febrile seizures. A pathogenic variant in the SCN1A gene is present in over 80% of cases.
Onset in childhood (age 2-12 years)[7][32]
Epilepsy with myoclonic-atonic seizures (EMAtS; formerly known as Doose syndrome):
Should be considered in pre-school children presenting with generalised seizures (generalised tonic-clonic, myoclonic, absence) who develop myoclonic-atonic seizures.
During a myoclonic-atonic seizure the child has a brief jerk (myoclonus), sometimes accompanied by an audible grunt, then abruptly loses muscle control and may fall to the ground. There is an associated loss of consciousness, but this is very brief (<3 seconds).
EEG is initially normal, although more commonly the inter-ictal recording shows some slowing with excess of theta activity. Appearance of the ictal EEG depends on the type of seizure: absences are associated with slow spike and waves, tonic-clonic seizures are associated with generalised 10- to 15-Hz poly-spikes, and myoclonic and atonic seizures are associated with irregular generalised spike-and-wave discharges. [Figure caption and citation for the preceding image starts]: Electroencephalogram (EEG): epilepsy with myoclonic-atonic seizuresCourtesy of Professor Eric Kossoff; used with permission [Citation ends].
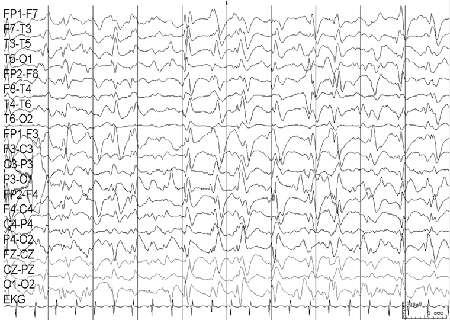
Most affected children have normal development before the onset of epilepsy, but about 50% go on to develop learning difficulties or mild/moderate intellectual disability.[34]
Lennox-Gastaut syndrome (LGS):
Onset is usually between ages 18 months and 8 years, with peak onset at 3-5 years.
The presence of tonic seizures lasting from 3 seconds to 2 minutes is mandatory for diagnosis.
Many other types of seizure may occur, including tonic-clonic, atonic, myoclonic, focal, and atypical absence.
Many patients show only some of the characteristic features over a certain period of time; clinical manifestations are often not all present in a given person.
EEG: abnormal inter-ictal EEG is a hallmark of this syndrome, showing diffuse, slow (<2.5 Hz) spike-and-wave complexes and generalised paroxysmal fast activity. [Figure caption and citation for the preceding image starts]: Electroencephalogram (EEG): Lennox-Gastaut syndromeCourtesy of Professor Eric Kossoff; used with permission [Citation ends].
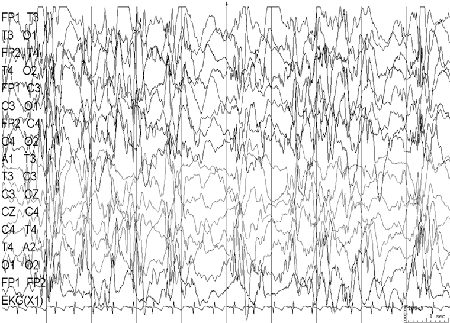
Most children have developmental delay.
Up to 75% of patients have recurrent episodes of non-convulsive status epilepticus, which can be difficult to recognise and treat.
Pathogenic variants in many genes, as well as a range of chromosomal abnormalities and copy number variants, have been associated with LGS.
Epilepsy with myoclonic absence (EMA):
Onset is typically around age 7 years (range 1-12 years).
Myoclonic absence seizures are the defining type of seizure; these last up to 1 minute and occur multiple times per day.
Tonic-clonic, clonic, atonic, or typical absence seizures may also occur.
Ictal EEG shows bilateral synchronous 3-Hz spike-wave discharges associated with 3-Hz myoclonic bursts; inter-ictal EEG has normal background activity, but up to 30% of patients have superimposed generalised spike-wave discharges.
Patients may have developmental impairment at presentation, and some will subsequently have a degree of intellectual disability.
Epilepsy with eyelid myoclonia (EEM):
Peak age at onset is between 6 and 8 years (range 2-14 years).
The most typical feature is eyelid myoclonia that occurs frequently with eye closure, may be provoked by light (photic induction), and is often associated with brief absence seizure. Seizures last only a few seconds but occur many times per day.
Other seizure types that may be seen include generalised tonic-clonic, myoclonic, and typical absence seizures.
EEG: on eye closure (or photic stimulation), brief discharges of fast activities are seen; a typical pattern is 3- to 6-Hz poly-spike waves.
Development is often normal, but intellectual disability may be seen, and is more likely in patients with prominent photic induction.
Childhood absence epilepsy (CAE):
Onset is typically between ages 4 and 10 years (range 2-13 years).
Typical absence seizures are the predominant seizure type.
Rarely, generalised tonic-clonic seizures can occur.
EEG background is normal, with paroxysms of 3-Hz generalised spike-wave. The characteristic pattern of the ictal recording is regular 3-Hz ictal generalised and symmetrical 3-Hz spike-wave. [Figure caption and citation for the preceding image starts]: Electroencephalogram (EEG): childhood absence epilepsy; shows typical 3 per second spike-and-wave patternCourtesy of Professor Eric Kossoff; used with permission [Citation ends].
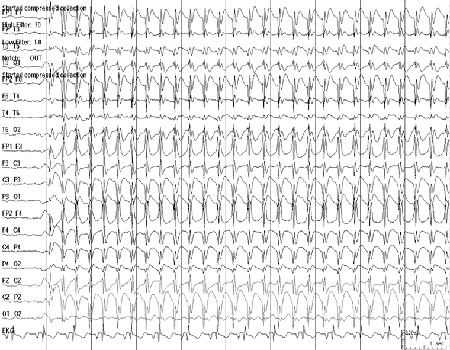
Development is typically normal.
Onset at a variable age
Onset of these syndromes commonly occurs in late childhood and adolescence.[7] Development is typically normal. Common comorbidities include mood disorders, ADHD, and learning disabilities.
Juvenile myoclonic epilepsy (JME):
Typical age at onset is between 10 and 24 years (range 8-40 years).
Myoclonic seizures are mandatory for diagnosis; they may be unilateral or bilateral, and occur almost exclusively in the morning and mainly in the upper limbs.
Generalised tonic-clonic seizures also occur in most patients, and absence seizures in about one third of cases.
Inter-ictal EEG shows 3.5- to 6-Hz spike- and poly-spike waves; most pronounced during sleep and drowsiness; rapid spikes followed by irregular slow waves are seen during myoclonus. [Figure caption and citation for the preceding image starts]: Electroencephalogram (EEG): juvenile myoclonic epilepsyCourtesy of Professor Eric Kossoff; used with permission [Citation ends].
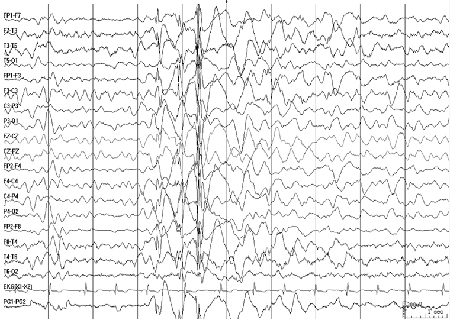
Juvenile absence epilepsy (JAE):
Typical age at onset is between 9 and 13 years (range 8-20 years).
The predominant seizure type is absence seizure. Absences tend to be longer and less frequent than the absences of childhood absence epilepsy, and loss of awareness is often less complete.
Generalised tonic-clonic seizures also occur in most patients.
Ictal EEG shows 3- to 5.5-Hz generalised spike-and-slow-wave discharges; inter-ictal EEG is usually normal.
Epilepsy with generalised tonic-clonic seizures alone (GTCA):
Typical age at onset is between 10 and 25 years (range 5-40 years).
Generalised tonic-clonic seizures are mandatory, and no other seizure types occur. Seizures often occur within 2 hours of waking, and are typically infrequent.
Inter-ictal EEG shows 3- to 5.5-Hz spike- and/or poly-spike-wave discharges.
Use of this content is subject to our disclaimer