Approach
Normal variation accounts for the majority of cases of short stature. Pathological causes are numerous but rare, and require comprehensive evaluation.[15]
A graded diagnostic approach, with a careful history, physical examination, and targeted laboratory and radiological evaluation is recommended in order to determine the aetiology and guide treatment. The first evaluation may not yield the diagnosis, and careful follow-up over months or even years is sometimes required. Meticulous measurements, accurate plotting, growth velocity derivations, and pubertal status assessments help the physician elucidate the cause.
After reaching a diagnosis, it is important to re-evaluate the patient periodically to confirm the diagnosis.[2][8][13][15][27][28]
History
A thorough history is important in establishing the cause of a child's short stature.[9][10]
Birth history
Birth weight, length, and gestation will identify whether the child was born small for gestational age.
Perinatal complications, such as hypoglycaemia or micropenis, are suggestive of growth hormone (GH) deficiency.
Medical history
Dyspnoea may suggest a cardiac or pulmonary cause, such as moderate/severe asthma, or chronic heart disease.
Diarrhoea may suggest coeliac disease or other malabsorption syndromes.
Blood in stools may indicate inflammatory bowel disease.
Joint pains and rash may indicate a systemic inflammatory condition such as juvenile idiopathic arthritis.
Recurrent respiratory infections with diarrhoea may raise suspicion of cystic fibrosis. Other symptoms include nasal polyps, delayed puberty, and faltering growth.
Patient may have diabetes mellitus with symptoms of polyuria, polydipsia, and weight loss.
Growth delay in the second year of life with a 'cherubic' appearance (looking much younger than stated age with a low muscle-to-fat ratio) and perinatal hypoglycaemia suggests GH deficiency. The growth pattern in GH insensitivity is often similar to that of GH deficiency, with extreme short stature.
There may be a history of headache or diplopia, suggestive of a craniopharyngioma with other pituitary hormone dysfunction, or there may be a history of recent brain surgery.[29] The child may be a long-term survivor of other malignancies such as acute lymphoblastic leukaemia, having received chemotherapy or radiotherapy.[30] Radiotherapy to the spine results in disproportionate short stature.[20]
Recent weight gain, acne, mood swings, and headaches may be present with Cushing's syndrome.
Fatigue, cold intolerance, dry skin, hair loss, constipation, lethargy, and weight gain suggest hypothyroidism.
Presence of multiple systemic congenital abnormalities (e.g., Turner syndrome, trisomy 21, DiGeorge syndrome) may raise suspicion of syndromic short stature.[31]
History of psychological abnormalities, bingeing, purging, and altered body image suggests anorexia nervosa or bulimia nervosa.
Family history
Elicit any history of chronic illnesses or antenatal ingestion of alcohol or other drugs.
Parental heights and calculation of the target height, and weights and heights of siblings, should be undertaken. A history of late pubertal development in either parent or of parental consanguinity should also be obtained.
Social history should ascertain family dynamics and raise any suspicion of neglect, abuse, or starvation. Parent-child bonding and interaction should be observed.
Dietary history
Adequate caloric intake and access to food should be ensured.
Medication history
Chronic corticosteroid treatment may lead to iatrogenic short stature.
Patients with diabetes mellitus may already be receiving insulin therapy.
Other medication history such as treatment for attention deficit-hyperactivity disorder should be obtained.
Development
May be delayed in syndromic short stature such as Prader-Willi syndrome, DiGeorge syndrome, and trisomy 21.
May also be delayed in severe cases of psychosocial deprivation.
Growth data
An examination of a child's growth over the years can provide important clues to the correct diagnosis. Accurate measurements, using correct age- and sex-specific growth charts, are mandatory to confirm that the child is short. Care must be taken to plot the height and weight based on the child's actual chronological age. Length should be measured supine in infants until 2 years of age, and standing thereafter.
Head circumference should be followed in children aged <3 years. Hydrocephalus may be present with achondroplasia or central nervous system (CNS) tumours.
Height should ideally be measured with a wall-mounted stadiometer while making sure that the child is shoeless, standing up straight, and not leaning on the wall, with his/her feet together and looking directly forwards. Infant length should be measured from crown of head to outstretched heel on a firm platform with a fixed headplate and a moveable footplate.
Growth charts
Clinicians in the US use growth charts that are developed by the Centers for Disease Control and Prevention (CDC). CDC: growth charts Opens in new window In 2006, the World Health Organization (WHO) released international growth charts for children aged 0 to 59 months. Similar to the 2000 CDC growth charts, these charts describe weight for age, length (or stature) for age, weight for length (or stature), and body mass index for age.
The 2006 WHO international growth charts are recommended for children aged <24 months. In the US, the CDC growth charts should continue to be used for the assessment of growth in people aged 2 to 19 years.[32]
WHO charts reflect growth patterns in breastfed children
The recommendation to use the 2006 WHO international growth charts for children aged <24 months is based on several considerations, including the recognition that breastfeeding is the recommended standard for infant feeding. Because breastfed infants demonstrate slower growth during 3 to 18 months of age compared with formula-fed infants, fewer children will be identified as underweight using the WHO charts compared with the CDC charts. However, gaining weight more rapidly than is indicated on the WHO charts may signal early signs of overweight.[Figure caption and citation for the preceding image starts]: The WHO and CDC growth chartsFrom the collection of Maria G Vogiatzi MD [Citation ends].
The UK Royal College of Paediatrics and Child Health has produced growth charts based on the WHO child growth standards. Royal College of Paediatrics and Child Health: UK-WHO growth charts, 0-18 years Opens in new window These UK-WHO growth charts are recommended for use in the 2017 guideline on faltering growth in children by the National Institute for Health and Care Excellence (NICE).[33]
Premature infants and disease-specific charts
Premature infants should have their growth plotted on charts designed to account for their gestational age (i.e., until 24 months for weight, 40 months for length, and 18 months for head circumference).
Disease-specific growth charts may also be used for syndromic short stature to compare growth with data from other children with the same pathology (e.g., Turner syndrome, achondroplasia, trisomy 21).[34] Special charts may be used to plot the upper and lower segments separately.
Proportionate short stature: involves both the torso and the lower extremities equally.
Disproportionate short stature: involves one segment more than the other. It generally suggests a diagnosis of skeletal dysplasia, such as achondroplasia (short lower segment), or a spinal disorder, such as previous spinal irradiation (short upper segment).[18][22]
Weight versus height
It is important to determine whether the child's weight or height is more affected.
Decreased or normal weight-to-height ratio suggests inadequate caloric intake or chronic illness as the aetiology for the short stature.
A high weight-to-height ratio suggests an endocrine aetiology, such as GH deficiency, GH insensitivity, hypothyroidism, or glucocorticoid excess, and should be referred to a paediatric endocrinologist.
Target height
Growth is strongly related to genetic potential. A child's target height is calculated as follows:
Girl = ([height of mother in inches + height of father in inches]/2) - 2.5 inches
Boy = ([height of mother in inches + height of father in inches]/2) + 2.5 inches.
A short child who is growing close to his/her target height percentile is likely to have familial short stature.[35] Growth deceleration during the first 2 years followed by a normal growth velocity, with acceleration late in adolescence, leading to a final height that is close to the target height suggests constitutional delay in growth and development.[Figure caption and citation for the preceding image starts]: Familial short stature (TH target height)From the collection of Maria G Vogiatzi MD [Citation ends].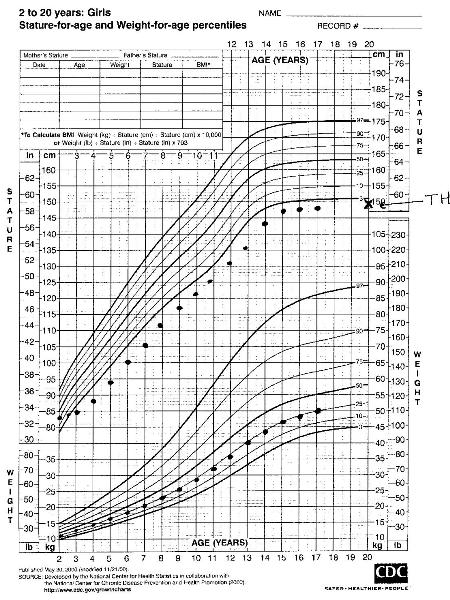 [Figure caption and citation for the preceding image starts]: Constitutional delay of growth and development (TH target height)From the collection of Maria G Vogiatzi MD [Citation ends].
[Figure caption and citation for the preceding image starts]: Constitutional delay of growth and development (TH target height)From the collection of Maria G Vogiatzi MD [Citation ends].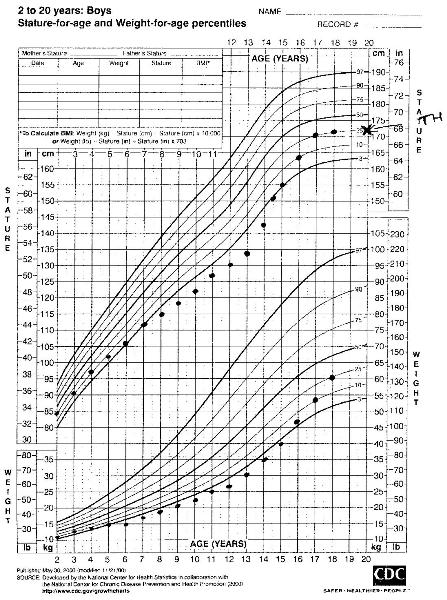
Growth velocity
During an evaluation of a child with short stature, it is important to have serial measurements plotted on a growth chart.
Growth velocity determines the change in height over time. It is calculated as the difference in height on 2 different occasions annualised over a year. Growth velocities depend on age and pubertal status.
Height that plots stably along a given percentile on the growth chart reflects normal growth velocity. Crossing percentiles in the downward or upward directions reflects abnormally low or accelerated growth velocities, respectively.[8][Figure caption and citation for the preceding image starts]: Severe growth hormone deficiency prior to treatment (TH target height)From the collection of Maria G Vogiatzi MD [Citation ends].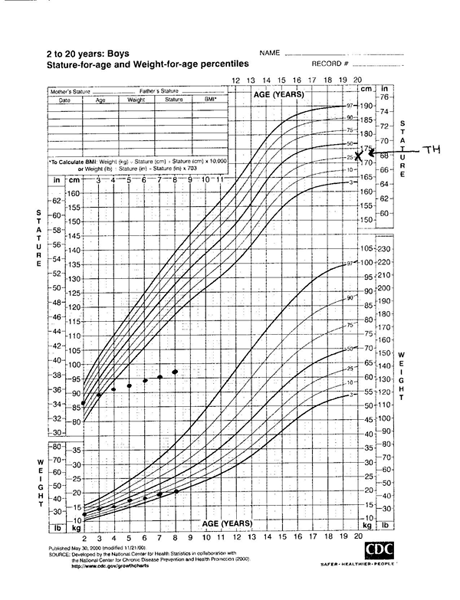
Physical examination
A complete review of systems needs to be undertaken in order to help exclude an undiagnosed syndrome or chronic medical condition.
Trisomy 21: hypotonia, upward slanting of palpebral fissures, round face, micrognathia, single palmar crease, brachycephaly
Turner syndrome: webbed neck, low hairline, broad chest, increased carrying angle, hypertelorism, posteriorly rotated ears
Russell-Silver syndrome: triangular face, asymmetry, clinodactyly
Prader-Willi syndrome: obesity, small hands and feet
DiGeorge syndrome: minor facial dysmorphism, cleft palate
Noonan syndrome: hypertelorism, low-set ears
Congenital GH deficiency: associated midline abnormalities such as cleft lip and palate, midfacial hypoplasia, single central incisor.
Vital signs:
Tachycardia may indicate cardiac failure; bradycardia may suggest eating disorders or severe hypothyroidism
Tachypnoea may be observed in respiratory disorders such as cystic fibrosis or other chronic cardiac and pulmonary diseases
Pallor may indicate anaemia due to chronic illnesses, malabsorption syndromes, or malignancies
Hypothermia may be observed in chronic hypothyroidism
Hypertension is present in Cushing syndrome.
General examination:[8][9][10]
Pallor, dry skin, facial coarsening, hair loss, non-pitting oedema suggest hypothyroidism
Lymphoedema is observed with Turner syndrome
Increased weight, cushingoid features (buffalo hump, hirsutism, violaceous striae) are present in Cushing syndrome; 'cherubic' appearance (looking much younger than stated age with a low muscle-to-fat ratio) suggests GH deficiency
Midline defects such as a single incisor or cleft palate may be found with congenital GH deficiency
Blue sclera and fractures are present in osteogenesis imperfecta
Characteristic bony deformities such as rachitic rosary or genu varum suggest rickets
Milk bottle caries, poor hygiene, or severe nappy rash suggest neglect; bruising in preambulatory infants or patterned bruising in older children suggest abuse, as do inadequately explained injuries; retinal haemorrhages may be present if the child has been shaken violently
Disproportionate short stature with shortening of the distal or proximal sections of upper or lower extremities is indicative of skeletal dysplasia; kyphoscoliosis or other spinal deformities are evident in children with achondroplasia, or there may be evidence of spinal surgeries
Muscle wasting, anaemia, and other signs of malnutrition suggest malabsorption, anorexia nervosa, bulimia nervosa, starvation, child abuse, neglect, or chronic severe medical conditions
Joint swellings, rash, murmur are present in juvenile idiopathic arthritis.
A murmur suggests congenital heart disease, isolated or associated with genetic syndromes
Signs of pneumonia may be present in cystic fibrosis and other respiratory conditions
Abdominal distension suggests coeliac disease and other malnutrition states
Abnormal neurology may be present with CNS tumours.
Pubertal examination
Pubertal status needs to be carefully assessed because it indicates skeletal maturation and growth potential. Pubertal delay points to a possibility of constitutional delay of growth and development, or hypogonadotrophic hypogonadism due to hypopituitarism.
Bone age assessment
Skeletal maturation is assessed by radiography. The appearance of representative epiphyseal centres on the x-ray is compared with age- and sex-appropriate published standards. The most commonly used method is that of Greulich and Pyle, which examines the left wrist and hand, but other methods such as the knee examination may be more helpful in infants. Bone age may also be used to predict final height, using the tables of Bayley and Pinneau.[36][37]
Many conditions that cause poor linear growth may also cause a delay in skeletal maturation, so the finding of maturation delay is not diagnostic of any particular condition.[12] Delayed bone age does, however, indicate that the short stature is partially reversible, because linear growth will continue until the epiphyses fuse.
Laboratory evaluation
Choice of tests should be based on the potential aetiologies after a careful history and physical examination.
First-line tests include:[9][10][38]
FBC
Complete metabolic profile (to screen for electrolyte abnormalities in renal and other chronic diseases) including looking for metabolic acidosis, and for hypocalcaemia (e.g., in DiGeorge syndrome, rickets)
Erythrocyte sedimentation rate to rule out underlying undiagnosed chronic inflammatory illness
Tissue transglutaminase to rule out coeliac disease
Urinalysis to rule out infection, haematuria, or proteinuria (with chronic renal disease), and to detect renal tubular acidosis
Thyroid function tests to screen for hypothyroidism
Insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 (IGFBP-3) to screen for GH deficiency
Karyotype in girls to rule out Turner syndrome.
Subsequent tests depend on the diagnosis and may include:
GH stimulation tests for GH deficiency; patients may also need magnetic resonance imaging of the brain and full pituitary evaluation to identify co-existing pathology.[39]
Diurnal cortisol levels, urinary cortisol levels, and a dexamethasone suppression test for Cushing syndrome
X-ray wrist and vitamin D levels for rickets; a skeletal survey in osteogenesis imperfecta, achondroplasia
An IGF-1 generation test in patients with a high basal GH and suspected to have GH insensitivity
Echocardiogram for congenital heart disease
Genetic evaluation with karyotyping for other genetic syndromes
Referral to a psychologist for eating disorders
Referral to the child protection team if there are concerns regarding abuse or neglect
Appropriate investigations for chronic illnesses such as a sweat test for cystic fibrosis, rheumatoid factor in juvenile idiopathic arthritis, HbA1c in diabetes mellitus, endoscopy in inflammatory bowel disease, and a referral to the appropriate specialist if underlying undiagnosed medical condition identified.
Diagnostic algorithm
A practical way to approach the differential diagnosis of pathological short stature is to divide patients into those with dysmorphic features and those without.[40]
Dysmorphic features are suggestive of genetic syndromes and may be disproportionate or proportionate based on the upper-to-lower segment ratio. Those who are not dysmorphic can be divided depending on the weight-to-height ratio.
The following algorithm will aid in narrowing the differential diagnosis. [Figure caption and citation for the preceding image starts]: Diagnostic algorithm of short statureFrom the collection of Maria G Vogiatzi MD [Citation ends].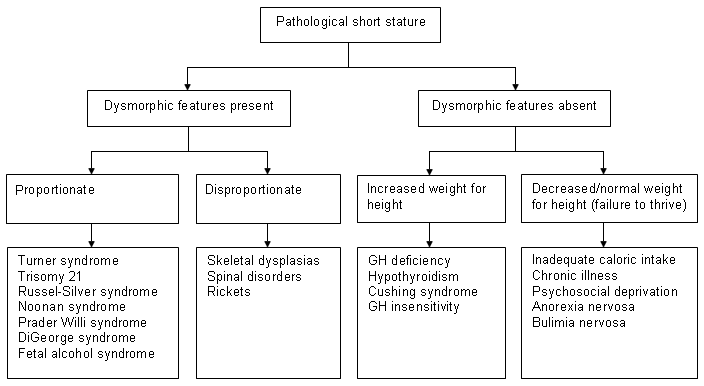
Use of this content is subject to our disclaimer