Approach
An infant with any degree of virilisation should be evaluated to determine the cause of genital differences. Patients with the most severe form of classical salt-wasting CAH present early in life with ambiguous genitalia in 46,XX individuals. 46,XY individuals or 46,XX with complete androgenisation/prader 5 genitalia may present with severe dehydration due to a salt-wasting crisis if not detected on the newborn screening test. The extent of virilisation in an affected female may be the same in simple virilising and in salt-wasting congenital adrenal hyperplasia (CAH).
While awaiting testing results, the infant should be monitored for signs of salt-wasting. If the cause is determined to be 21-hydroxylase deficiency (21-OHD), treatment should commence.
The salient features of classical simple-virilising CAH are antenatal virilisation in 46,XX individuals and progressive postnatal signs of androgen excess (i.e., with accelerated growth and advanced bone ages, premature adrenarche, precocious puberty) without evidence of mineralocorticoid deficiency.
The diagnosis of CAH should also be considered in infants who present with poor weight gain, poor feeding, and dehydration with hyponatraemia and hyperkalaemia.[2]
Antenatal diagnosis can be performed when both parents are carriers of CYP21A2 mutations; most often this situation arises when they have a previous child with 21-OHD.[11]
Antenatal diagnosis can be performed in the first trimester by molecular genetic analysis of fetal DNA from chorionic villus sampling or amniocentesis
Non-invasive sampling of free fetal DNA has also been developed.[12]
Clinical evaluation
Classical salt-wasting CAH
Diagnosis of a 46,XX individual is usually made at birth due to genital atypia. Common findings include clitoral enlargement and partially fused labia majora. In addition, the distal part of urethra and vaginal canal may fuse to form a urogenital sinus, which presents as a single perineal opening instead of two distinct urethra and vaginal orifices.[2]
Differentiation of the external genitalia is unaffected in newborn males. Diagnosis at birth usually depends on antenatal or newborn screening.
In addition to symptoms of excess androgen, infants present with renal salt-wasting. Symptoms include poor feeding, weight loss, failure to thrive, vomiting, dehydration, hypotension, hyponatraemia, and hyperkalaemic metabolic acidosis progressing to adrenal crisis (azotaemia, vascular collapse, shock, and death). Adrenal crisis typically occurs at 1 to 2 weeks of age. Early diagnosis by newborn screening with appropriate treatment have significantly decreased the salt-wasting crisis in the newborn and associated mortality.
While family history may reveal an affected relative, this is not necessary or common as this is a recessively inherited condition.
Signs of hyperandrogenism in affected children may include early onset of facial, axillary, and pubic hair; adult body odour; and rapid somatic growth. The peripheral hyperandrogenism may also activate true central precocious puberty. The early growth spurt is accompanied by premature epiphyseal maturation and closure, resulting in a final height that is below that expected from parental heights. Patients tend to be tall children, but short adults.
In adolescence and adult age, signs of hyperandrogenism in females may include temporal balding, severe acne, irregular menses, hirsutism, and infertility if not adequately treated.
Menstrual irregularity and secondary amenorrhoea as well as primary amenorrhoea can occur in those with poor hormonal control. However, with optimal suppression of adrenal hyperandrogenism, fertility is preserved and woman can enjoy spontaneous pregnancies leading to healthy offspring.[13]
Males may experience development of testicular adrenal rest tumours (TART), which are benign but can cause obstructive azoospermia.[2] TARTs can occur bilaterally from a young age and can be detected by routine testicular sonograms. Leydig cell dysfunction and oligospermia, regardless of presence of TARTs, have been described in cases of poor adrenal control.
At all ages, individuals are at risk for adrenal crisis with major physiological stress, such as infection, severe injury or a surgical procedure. Adrenal crisis can start with vomiting and rapidly lead to haemodynamic instability and hypoglycaemia. Adrenal crisis has been reported as a leading cause of death in classical CAH. Stress doses of glucocorticoids are prescribed during physical stress to prevent adrenal crisis.
Classical simple-virilising CAH
Presentation and symptoms during infancy, childhood, adolescence and adulthood are the same as in the salt-wasting type. However, individuals with simple-virilising CAH have adequate aldosterone secretion that prevents salt-wasting crisis in infancy.
Most 46,XY individuals are identified by newborn screening. Otherwise, they may present later on in life with signs related to adrenal androgen excess.
Non-classical CAH
A positive family history may be present
The phenotype can be variable and many individuals may not come into medical attention
Genital development is normal. Some females may experience mild clitoromegaly.
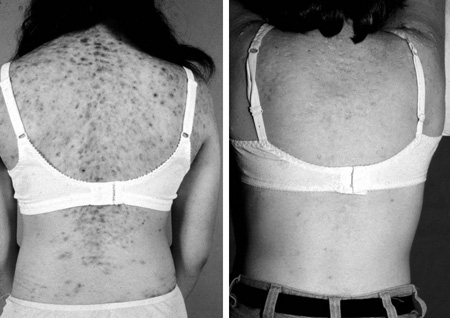
Symptoms can present during childhood and may include acne, premature development of pubic hair, early puberty, accelerated growth, advanced bone age, and reduced adult stature as a result of premature epiphyseal fusion. [Figure caption and citation for the preceding image starts]: Skin before and after treatment with dexamethasone for 3 monthsPermission granted by The Endocrine Society [Citation ends].
Adolescent girls and women may present with complains of hirsutism, temporal baldness, delayed menarche or menstrual irregularities.[Figure caption and citation for the preceding image starts]: Male-pattern baldness typical of females affected with non-classical 21-hydroxylase deficiencyPermission granted by The Endocrine Society [Citation ends].

Hormonal tests
The diagnosis of 21-OHD is confirmed by hormonal evaluation. A very high concentration of 17-hydroxyprogesterone (17-OHP), the steroid precursor that accumulates in 21-OHD, is diagnostic of classical disease.
17-OHP can also be elevated in healthy neonates during the first 1 to 2 days of life and in premature and ill neonates.[2]
While 17-OHP levels are often diagnostic in the classical form, values may be insufficiently elevated in non-classical disease. False-positive and false-negative results are common if samples are obtained immediately after birth.[14] In such cases, the adrenocorticotrophic hormone (ACTH) stimulation test is crucial in establishing the diagnosis of the non-classical form. During ACTH testing, 17-OHP and delta-4-androstenedione levels are measured at baseline and at 60 minutes following an intravenous injection of cosyntropin. These values can then be plotted on a published nomogram to ascertain disease severity.[15] Full adrenal profile should be obtained after the rapid ACTH stimulation test.[14]
In individuals with borderline 17-OHP levels, we recommend obtaining a measurement of additional steroids, such as 11-deoxycorticosterone, dehydroepiandrosterone (DHEA) and 17-OHP, after a cosyntropin stimulation test, to differentiate 21-OHD from other rare forms of CAH. Finally, measurement of cortisol reveals the presence or absence of adrenal sufficiency (specifically with regard to non-classical CAH).[1]
In infant and newborn screening, blood is taken by heel prick and blotted on microfilter paper for the measurement of 17-hydroxyprogesterone. A 26-year longitudinal nationwide study of neonatal screening for CAH in Sweden showed that screening is highly effective in detecting the salt-wasting form. The sensitivity of newborn screening is less in the simple virilising and non-classical forms.[16]
Other tests
Newborns or infants with ambiguous genitalia are recommended for karyotyping or fluorescence in situ hybridisation (FISH) for X and Y chromosome detection, and an ultrasound of the pelvis and adrenal glands to identify associated abnormalities.[2]
Serum chemistries
Serum chemistries are useful for newborns (after the first day of life) and for infants with atypical genitalia or at risk for 21-OHD CAH.[2] In a salt-wasting crisis, results often indicate severe dehydration, hyponatraemia, hyperkalaemia, and metabolic acidosis; however, this would not be expected until 1 to 2 weeks of life.[17]
Plasma renin activity
Plasma renin activity is inversely related to intravascular volume status. In volume-depleted states, such as salt-wasting in classical CAH, the values are elevated in variable degrees. Plasma renin activity measurement is recommended in patients suspected of classical salt-wasting CAH.[14] Unfortunately, a high plasma renin in untreated CAH is not diagnostic of salt-wasting CAH as 17-OHP, at high concentrations, has mineralocorticoid receptor antagonistic properties. Thus, high 17-OHP increases the plasma renin.[17] Renin may also be elevated in states of volume depletion, heart failure and other critical illness. The use of steroid medicines can lower the renin level. High plasma renin activity levels indicate aldosterone deficiency and potential hypovolaemia.
Genetic analysis
If there is a family history of the disease or one of the future parents has the disease, genetic analysis is strongly recommended. Both parents should also have genetic testing to determine if they carry a mutation. Genotyping may also be helpful if the results of the adrenocortical profile after a rapid ACTH stimulation test are equivocal.[14]
Gene analysis of CYP21A2 that detects the most common mutations is established in commercially available laboratories. Genetic analysis can confirm the diagnosis and is important for antenatal counselling and family planning. Studies report variable genotype-phenotype concordance, from 50% to over 97%.[9][10]
In a study of 213 patients with 21-OHD, genotype accurately predicted phenotype in 85.1 % of patients with simple-virilising CAH, 90.5% of patients with salt-wasting CAH, and 97.8% of patients with non-classical CAH.[10] However, in a subsequent study of 1507 patients with 21-OHD, genotype correlated with phenotype in 46.7% of cases, with the strongest correlation in the salt-wasting and non-classical forms.[9]
Use of this content is subject to our disclaimer