History and exam
Key diagnostic factors
common
presence of risk factors
Key risk factors include family history of joint hypermobility or EDS, and genetic mutations.
Pattern of inheritance is usually autosomal dominant, although the rare subtypes are autosomal recessive (kyphoscoliotic and dermatosparaxis EDS).
joint hypermobility
A feature of all subtypes of EDS. Usually less obvious in vascular EDS, except in the hands.
Presence of joint hypermobility can be established directly by the Beighton 9-point score or indirectly using the 5-question questionnaire.[16][17]
Hypermobility should be sought in joints outside the 5 sites that form part of the Beighton scoring system, as each hypermobile joint identified adds evidence of joint hypermobility.[Figure caption and citation for the preceding image starts]: Joint hypermobility demonstrated by opposition of thumb to volar aspect of forearmFrom the collection of Dr Rodney Grahame; used with permission [Citation ends].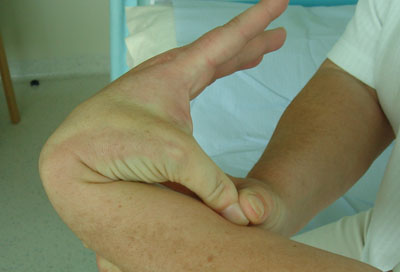 [Figure caption and citation for the preceding image starts]: Joint hypermobility demonstrated by hyperextension of the elbow to >90°From the collection of Dr Rodney Grahame; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Joint hypermobility demonstrated by hyperextension of the elbow to >90°From the collection of Dr Rodney Grahame; used with permission [Citation ends].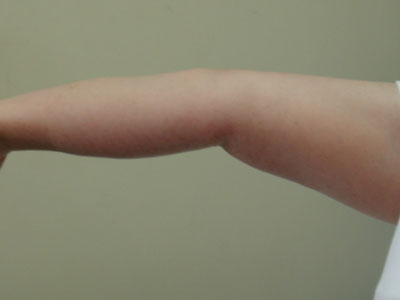 [Figure caption and citation for the preceding image starts]: Joint hypermobility demonstrated by hyperextension of the kneeFrom the collection of Dr Rodney Grahame; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Joint hypermobility demonstrated by hyperextension of the kneeFrom the collection of Dr Rodney Grahame; used with permission [Citation ends].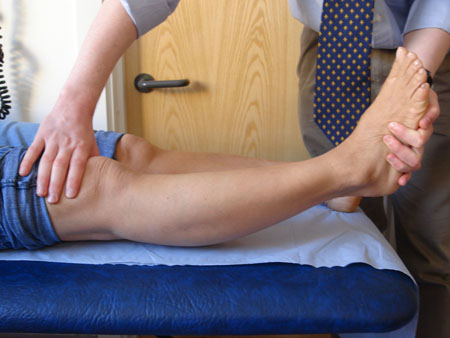 [Figure caption and citation for the preceding image starts]: Joint hypermobility demonstrated by placing hands flat on floor with knees fully extendedFrom the collection of Dr Rodney Grahame; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Joint hypermobility demonstrated by placing hands flat on floor with knees fully extendedFrom the collection of Dr Rodney Grahame; used with permission [Citation ends].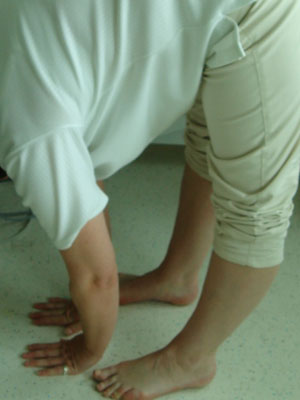

A video demonstration of Beighton score in a hypermobile Ehlers-Danlos syndrome patient.
joint or spine pain
Older patients usually present with multiple-site joint or spinal pain, often brought on by unaccustomed, though not necessarily excessive, physical activity and in the absence of signs of inflammation.
Unlike inflammatory joint disease (IJD), there is no visible joint swelling, warmth, or redness, and no complaint of early morning stiffness unless there is an associated tendonitis. Contrary to what is seen in IJD, the joints move well and, despite pain, may retain their hypermobility.
motor delay in infancy
History of delayed walking (age >18 months), often with omission of crawling and/or substitution of bottom-shuffling, is common in hypermobile infants. Once walking is initiated, the motor delay resolves.
chronic pain syndrome
Characterised by progressively severe chronic pain, often with fibromyalgia-like tender points on palpation, and accompanied by severe fatigue, autonomic dysfunction, and psychosocial morbidity (anxiety, depression, phobias). A key element is kinesophobia, avoiding movement as a means of avoiding pain.
Seen in about one quarter of patients attending hypermobility clinics.[22]
Usually occurs due to sudden injury (e.g., whiplash injury resulting from a significant motor vehicle accident) or unusual physical activity during home improvement, sporting activities, or over demanding work activities.
fatigue
Chronic fatigue is often associated with chronic pain in patients with hypermobile EDS.
Hypermobile children tire easily and often want to be carried.
recurrent joint dislocation or subluxation
Because ligaments are lax, joints are unstable and dislocate or sublux easily and repeatedly, in some cases several times a day.
muscle pain and/or muscle spasm
Tender (or non-tender) muscle spasm can often be palpated in the neck and paravertebral muscles, and sometimes in the muscles surrounding particularly unstable and/or painful joints.[21]
Hypermobile children may have joint and/or muscle pain after exercise (growing pains).
soft, silky skin texture
Skin has a characteristic soft and silky feel. Usually the only skin finding associated with hypermobile EDS.
semi-transparent skin
Skin may have easily visible underlying tendons and veins.[Figure caption and citation for the preceding image starts]: Thin translucent skinBMJ 2019;366:l4966 [Citation ends].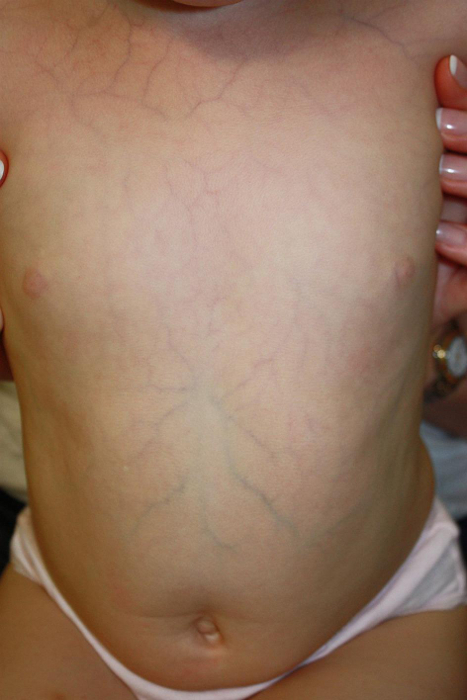
thin and stretchy double fold of skin
A double fold of skin picked up on the dorsum of the hand is often felt to be thin and, when lifted away from the dorsum of the hand, shows more stretchiness than skin of people without EDS. [Figure caption and citation for the preceding image starts]: Increased skin stretchiness demonstrated; common finding in EDSFrom the collection of Dr Rodney Grahame; used with permission [Citation ends].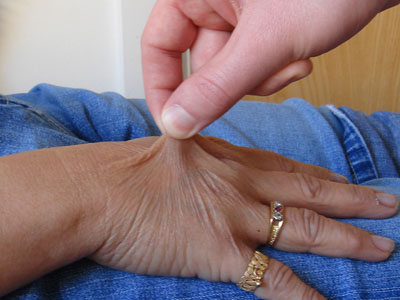
atrophic scars
Scars (from previous surgery, lacerations, abrasions, chicken pox, or BCG immunisation) have less collagen and are, therefore, usually thin in depth and wide in breadth. Typically they wrinkle when squeezed between the examiner's index finger and thumb.[Figure caption and citation for the preceding image starts]: Atrophic scarringBMJ 2019;366:l4966 [Citation ends].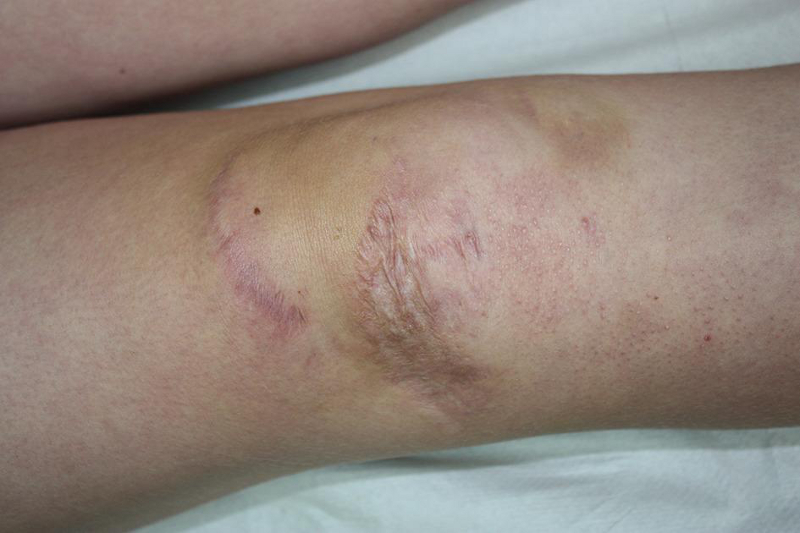
easy bruising
Patient often notices bruising but does not recall any precipitating injury.
stretch marks
Characteristically, stretch marks (striae atrophicae) appear during maximal adolescent growth (between the ages of 11 and 13 years) on thighs, loins, breasts, and, occasionally, shoulders and knees.
poor wound healing and/or wound dehiscence
Wound healing (involves the laying down of scar tissue or collagen) is delayed and may be incomplete. Wound dehiscence can occur due to fragility of the soft tissues.
significant injury
Because the tissues are more fragile than normal, they are at particular risk of traumatic or overuse injury. Therefore, injuries that would be inadequate to damage ligament, tendon, muscle, bone, or skin in normal people may result in significant damage.
history of delayed onset of local anaesthesia
Apparent resistance to the effects of local anaesthetics is seen in about two-thirds of patients.[18]
Other diagnostic factors
common
muscle hypotonia
Muscles may feel doughy on palpation.
varicose veins
Suggests weakness of supporting connective tissue.
abdominal wall, inguinal, or para-umbilical hernia
Suggests weakness of supporting connective tissue.
uterine or rectal prolapse
Suggests weakness of supporting connective tissue.
orthostatic hypotension
A sustained drop in blood pressure >20 mmHg within 3 minutes of standing.
orthostatic intolerance
Development of symptoms within 10 minutes of upright posture that improve upon lying down.
postural orthostatic tachycardia syndrome
Greater than 30-bpm increase in pulse on standing or >120 bpm within 10 minutes of head-up tilt-table testing, both in the absence of orthostatic hypotension that might trigger a normal tachycardic response.[26][27]
Symptoms that occur with standing include light-headedness, palpitations, tremors, generalised weakness, blurry vision, exercise intolerance, and fatigue.
neurally mediated hypotension
Can occur in susceptible individuals subsequent to orthostatic stress, emotional stress, urination, coughing, swallowing, and physical exercise. Causes a >25 mmHg drop in systolic blood pressure when standing.
Patients have daily light-headedness and other symptoms, but learn to adapt to these symptoms to reduce morbidity.
marfanoid habitus
May be present in association with EDS (usually incomplete). Common finding in hypermobile and kyphoscoliotic EDS, but is not necessary for diagnosis.
Features include: high arched palate; arachnodactyly; pectus excavatum or carinatum; scoliosis; arm span to height ratio >1.05; tall stature with reduced upper segment to lower segment (US/LS) ratio of <0.85; foot length (heel to first toe) to height ratio >0.15; hand length (wrist crease to third finger) to height ratio >0.11.[23]
gastrointestinal manifestations
Patients may have signs and symptoms suggesting a gastrointestinal disorder, such as gastritis/GORD (heartburn and acid regurgitation), gastroparesis (chronic nausea, vomiting, epigastric pain, bloating, and early satiety), irritable bowel syndrome (recurrent abdominal pain or discomfort associated with a change in stool frequency or form; pain or discomfort may be relieved by defecation), or rectal evacuatory disorder (characterised by difficulty with defecation, with or without need for manual evacuation and laxatives).[28][29]
gynaecological manifestations
Most women with hypermobility EDS experience significant gynaecological symptoms. Complaints include abnormal bleeding, dysmenorrhoea, and dyspareunia. Some also report a diagnosis of endometriosis, but systematic studies are lacking. There may be a higher rate of spontaneous miscarriages. Hormones may play a role in modifying symptom severity.[30]
uncommon
eye abnormalities
Suggests weakness of supporting connective tissue.
Lids appear drooping, but this does not extend to ptosis.
Anti-mongoloid slant is a condition in which the nasal corners of the palpebral fissure are higher than the temporal corners, as opposed to the typical mongoloid slant.
Blue sclera is due to thinning of the sclera. It is a non-specific feature of collagen deficiency.
Myopia is common in hypermobility syndromes.
mid-systolic click or late systolic murmur
May suggest mitral valve prolapse due to weak, lax, and less-effective connective tissues.
Risk factors
strong
family history of joint hypermobility or EDS
In cases of hypermobile EDS (hEDS) the pattern of inheritance is autosomal dominant, so that 50% of offspring of an affected person would be expected to inherit the mutated gene and develop the phenotype.[13] Classical, vascular, arthrochalasia, and periodontal subtypes are also autosomal dominant in nature, whereas the other subtypes are inherited in an autosomal recessive manner.
Studies show that the heritability factor (the proportion of phenotypic variation in a population that is attributable to genetic variation among people) of joint hypermobility is >70%.[14]
Most affected people do not develop symptoms at all, or they develop only minor symptoms during their lifetime.
genetic mutations
EDS (in keeping with other heritable disorders of connective tissues) is caused by one or more genetic aberrations affecting genes encoding for, or modifying, connective tissue proteins, such as collagen and matrix proteins (e.g., tenascin). Specific genetic aberrations lead to specific risks, such as severe skin pathology in classical EDS and vascular collapse due to arteriovenous rupture in vascular EDS.[12]
In some cases the gene product modifies a connective tissue protein (e.g., kyphoscoliotic and dermatosparaxis EDS).
The genetic aetiology is not known in hEDS.
Use of this content is subject to our disclaimer