Wilson's disease is uncommon and the diagnosis is often missed. This is due to its systemic involvement and varied clinical presentations with overlapping features. The mean delay from symptom onset to diagnosis is 2 years.[20]Shribman S, Warner TT, Dooley JS. Clinical presentations of Wilson disease. Ann Transl Med. 2019 Apr;7(suppl 2):S60.
https://atm.amegroups.com/article/view/25358/23684
http://www.ncbi.nlm.nih.gov/pubmed/31179297?tool=bestpractice.com
Wilson's disease can manifest as liver disease (with jaundice, and possibly ascites and oedema) or as a neurological movement disorder (with tremor, dystonia, rigidity), or it can be diagnosed at the asymptomatic stage.[21]Brewer GJ. Recognition, diagnosis, and management of Wilson's disease. Proc Soc Exp Biol Med. 2000 Jan;223(1):39-46.
http://www.ncbi.nlm.nih.gov/pubmed/10632959?tool=bestpractice.com
[22]Starosta-Rubinstein S, Young AB, Kluin K, et al. Clinical assessment of 31 patients with Wilson's disease: correlations with structural changes on magnetic resonance imaging. Arch Neurol. 1987 Apr;44(4):365-70.
http://www.ncbi.nlm.nih.gov/pubmed/3827691?tool=bestpractice.com
[23]Brewer GJ. Novel therapeutic approaches to the treatment of Wilson's disease. Exp Opin Pharmacother. 2006 Feb;7(3):317-24.
http://www.ncbi.nlm.nih.gov/pubmed/16448326?tool=bestpractice.com
Early diagnosis is important because therapy is effective in achieving copper balance. Symptoms of the disease can be prevented or reversed if treated early, and thus family members of diagnosed patients should be screened.
Hepatic presentation
A patient presenting with liver disease secondary to Wilson's disease may have a history of unexplained hepatitis or aminotransferase elevations, thrombocytopenia, pancytopenia, indirect hyperbilirubinaemia, or Coombs-negative intravascular haemolysis.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[20]Shribman S, Warner TT, Dooley JS. Clinical presentations of Wilson disease. Ann Transl Med. 2019 Apr;7(suppl 2):S60.
https://atm.amegroups.com/article/view/25358/23684
http://www.ncbi.nlm.nih.gov/pubmed/31179297?tool=bestpractice.com
[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
Alternative presentations in adults and children include acute hepatitis, fatty liver, acute liver failure, or cirrhosis (compensated or decompensated).[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
Up to 20% of people with Wilson's disease present with acute liver failure, with symptoms of jaundice, hepatic encephalopathy, coagulopathy, ascites, and renal failure.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
Children usually present after age 5 years and may be asymptomatic, with hepatomegaly or abnormal serum transaminases detected incidentally.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
Patients may initially receive a diagnosis of acute viral hepatitis, metabolic dysfunction-associated steatotic liver disease (previously known as non-alcoholic fatty liver disease), or autoimmune hepatitis.[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
Physical examination may reveal hepatomegaly or splenomegaly. There may be signs of chronic liver disease, such as spider angiomata, gynaecomastia, ascites, peripheral oedema, easy bruising, jaundice, or encephalopathy. The patient might have Kayser-Fleischer (KF) rings or sunflower cataracts (deposits of copper in the lens).[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
All patients with liver abnormalities of uncertain cause should be screened for Wilson's disease.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Neurological presentation
A patient with neurological disease may have a history of symptoms of a movement disorder, including tremor, incoordination, sloppy handwriting, dysarthria, muscle stiffness, rigidity, postural abnormality, gait abnormality, chorea/athetosis, seizures, or drooling.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
[26]Taly AB, Meenakshi-Sundaram S, Sinha S, et al. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine (Baltimore). 2007 Mar;86(2):112-21.
https://journals.lww.com/md-journal/Fulltext/2007/03000/Wilson_Disease__Description_of_282_Patients.6.aspx
http://www.ncbi.nlm.nih.gov/pubmed/17435591?tool=bestpractice.com
Neuropsychiatric symptoms usually develop in the second or third decade of life, but they have been reported in children under 10 years.[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
One or more psychiatric condition may be present at a time.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Patients often have abnormal behaviour, personality change, depression, psychosis, bipolar disorder, and cognitive impairment.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[27]Dening TR, Berrios GE. Wilson's disease. Psychiatric symptoms in 195 cases. Arch Gen Psychiatry. 1989 Dec;46(12):1126-34.
http://www.ncbi.nlm.nih.gov/pubmed/2589927?tool=bestpractice.com
[28]Akil M, Brewer GJ. Psychiatric and behavioral abnormalities in Wilson's disease. Adv Neurol. 1995;65:171-8.
http://www.ncbi.nlm.nih.gov/pubmed/7872138?tool=bestpractice.com
Other behavioural abnormalities that may be present are temper tantrums, anxiety, loss of memory, inability to focus on tasks, impulsiveness, and disinhibition.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
[28]Akil M, Brewer GJ. Psychiatric and behavioral abnormalities in Wilson's disease. Adv Neurol. 1995;65:171-8.
http://www.ncbi.nlm.nih.gov/pubmed/7872138?tool=bestpractice.com
Patients can be mistaken for having Parkinson's disease or multiple sclerosis (although there is no sensory deficit).
On physical examination of the patient presenting with neurological disease, the following may be noted.
Tremor (of any type).
Dystonia and rigidity of any part of the body but most often the hands. This can cause postural and gait abnormalities. A dystonic smile may be seen.
Dysdiadochokinesis and evidence of clumsiness or lack of coordination.
Handwriting is often sloppy, but may be small (micrographia).
Speech may be abnormal, but the abnormalities are not specific for Wilson's disease. Words may be slurred and they may be of low volume (hypophonia). It may be almost stuttering in type (echolalia).
Dysphagia may cause the patient to drool and have difficulty with muscles of the lips and face.
Extra-ocular movements are occasionally abnormal.
Sensation is normal.
KF rings may be apparent (but should always be confirmed by slit-lamp examination). Sunflower cataracts may also be seen. [Figure caption and citation for the preceding image starts]: Eye demonstrating Kayser-Fleischer ringAdapted from BMJ (2009); used with permission [Citation ends].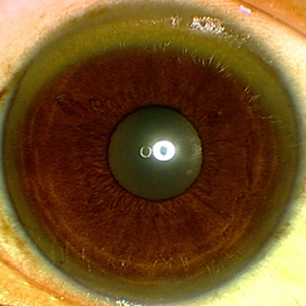
Investigations
A detailed personal and family history should be collected from patients suspected to have Wilson's disease.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
All patients should be investigated with liver function tests, slit-lamp examination for KF rings, serum ceruloplasmin, and 24-hour urinary copper measurement.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Further investigations may be indicated depending on the clinical presentation and results of the initial tests. No test is capable of diagnosing Wilson's disease in isolation.[29]Ryan A, Nevitt SJ, Tuohy O, et al. Biomarkers for diagnosis of Wilson's disease. Cochrane Database Syst Rev. 2019 Nov 19;(11):CD012267.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD012267.pub2/full
http://www.ncbi.nlm.nih.gov/pubmed/31743430?tool=bestpractice.com
The Leipzig score compiles clinical features and investigation results and calculates a score. A score of 4 or more is diagnostic for Wilson's disease.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[6]Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003 Jun;23(3):139-42.
http://www.ncbi.nlm.nih.gov/pubmed/12955875?tool=bestpractice.com
See Diagnostic criteria.
Liver function tests, including prothrombin time
Liver function tests (LFTs) are almost always abnormal in patients with hepatic presentation.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are present in 40% to 60% of patients at presentation.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
Approximately 12% of patients have elevated bilirubin, due to liver injury, Coombs-negative haemolysis, or both.[6]Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003 Jun;23(3):139-42.
http://www.ncbi.nlm.nih.gov/pubmed/12955875?tool=bestpractice.com
[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
Those with acute liver failure may have low albumin and prolonged international normalised ratio (INR). Those with decompensated cirrhosis may have prolonged INR, thrombocytopenia, mixed pattern of LFTs, and low albumin.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
In patients with neurological presentation, ALT and AST may also be abnormal because there can be overlap with liver involvement.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
24-hour urinary copper measurement
The sample should be collected in a container free of trace elements. Twenty-four-hour urinary copper >100 micrograms is consistent with Wilson's disease.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Twenty-four hour urinary copper >40 micrograms may suggest Wilson's disease and require further investigation.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
Urinary copper excretion can also be high in cholestatic and autoimmune conditions or protein-losing enteropathy, so diagnosis of the disease should not be made on urine copper values alone.
Slit-lamp examination for KF rings
KF rings are gold-brown pigments representing copper deposition in the membrane of Descemet of the cornea. They usually appear in superior and inferior portions of the cornea and then become circumferential as copper deposition progresses.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
KF rings are present in 95% of patients with neurological presentations and in 44% to 62% of patients with hepatic presentations.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
They can disappear with adequate treatment for Wilson's disease.
Serum ceruloplasmin and non-ceruloplasmin-bound copper concentration (NCC)
Ceruloplasmin, synthesised in the liver, is the major copper-carrying protein in the blood. Normal serum ceruloplasmin level is 200-350 mg/L (20-35 mg/dL). A serum ceruloplasmin level of <50 mg/L (<5 mg/dL) strongly suggests Wilson's disease.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Further testing is required to make the diagnosis.
Serum ceruloplasmin is not always diagnostic because approximately 20% of patients with Wilson's disease have results within normal range.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
Around 1% of the population are heterozygous for an ATP7B mutation and have an intermediate low ceruloplasmin level. Therefore, a normal serum ceruloplasmin does not exclude a diagnosis of Wilson's disease.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Serum ceruloplasmin <200 mg/L (<20 mg/dL) has a sensitivity of 77.1% to 99% and a specificity of 55.9% to 82.8% for diagnosis of Wilson's disease. Serum ceruloplasmin <100 mg/L (<10 mg/dL) has a sensitivity of 65.7% to 94.4%, and a specificity of 96.6% to 100%, for a diagnosis of Wilson's disease.[29]Ryan A, Nevitt SJ, Tuohy O, et al. Biomarkers for diagnosis of Wilson's disease. Cochrane Database Syst Rev. 2019 Nov 19;(11):CD012267.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD012267.pub2/full
http://www.ncbi.nlm.nih.gov/pubmed/31743430?tool=bestpractice.com
Assay by the enzymatic method seems most accurate, but antibody-mediated assay can be used as an alternative. Ceruloplasmin can be increased in cases of inflammation as it is an acute phase reactant.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
It can also be increased in pregnancy and oestrogen intake. It is usually low in infancy and is usually not measured until the child is >6 months.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Ceruloplasmin can also be low in cases of protein-losing enteropathy, non-selective renal protein loss, end-stage liver disease, nephrotic syndrome, neurological disorders (cervical dystonia), absolute copper deficiency (improper formulation of total parenteral nutrition omitting copper, after gastric or bariatric surgery, or chronic ingestion of zinc in excess), Menkes' disease, aceruloplasminaemia, MEDNIK syndrome, AP1B1 disorder, congenital glycosylation disorder (PGM1‐CDG, CCDC115‐CGD, or TMEM119‐CDG), or Niemann‐Pick's disease type C.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
[30]Merle U, Eisenbach C, Weiss KH, et al. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson's disease. J Hepatol. 2009 Nov;51(5):925-30.
http://www.ncbi.nlm.nih.gov/pubmed/19720421?tool=bestpractice.com
Direct assay of free copper has been suggested to help in diagnosis.[31]McMillin GA, Travis JJ, Hunt JW. Direct measurement of free copper in serum or plasma ultrafiltrate. Am J Clin Pathol. 2009 Feb;131(2):160-5.
https://academic.oup.com/ajcp/article/131/2/160/1765310
http://www.ncbi.nlm.nih.gov/pubmed/19141375?tool=bestpractice.com
Serum copper is usually low in Wilson's disease due to decrease in circulating ceruloplasmin. The non-ceruloplasmin-bound copper concentration (NCC) can be calculated using the following equation:[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
NCC = serum copper concentration (micrograms/dL) - (3.15 x serum ceruloplasmin concentration (mg/dL)).
A level greater than 25 micrograms/dL is often seen in untreated patients.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Elevated levels of NCC can be seen in cholestasis or excessive copper ingestion. Although often useful, the NCC cannot be interpreted if serum ceruloplasmin has been overestimated, because it may be a negative number.[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Liver biopsy
If the diagnosis is uncertain, the definitive diagnostic test is a liver biopsy with quantitative assay of copper (normal is 20 to 50 micrograms/g dry weight of tissue; in Wilson's disease the level is >250 micrograms/g).[5]Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2023 Apr 1;77(4):1428-55.
https://journals.lww.com/hep/fulltext/2023/04000/a_multidisciplinary_approach_to_the_diagnosis_and.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/36152019?tool=bestpractice.com
Absence of copper stain on the liver tissue does not rule out Wilson's disease due to sporadic deposition of copper within the hepatocytes.[32]Stromeyer FW, Ishak KG. Histology of the liver in Wilson's disease: a study of 34 cases. Am J Clin Pathol. 1980 Jan;73(1):12-24.
http://www.ncbi.nlm.nih.gov/pubmed/7352414?tool=bestpractice.com
[33]Johncilla M, Mitchell KA. Pathology of the liver in copper overload. Semin Liver Dis. 2011 Aug;31(3):239-44.
http://www.ncbi.nlm.nih.gov/pubmed/21901654?tool=bestpractice.com
Imaging
MRI of the brain is useful in neurological presentation. The most common findings are:[16]Dusek P, Litwin T, Członkowska A. Neurologic impairment in Wilson disease. Ann Transl Med. 2019 Apr;7(suppl 2):S64.
https://atm.amegroups.com/article/view/24752/23688
http://www.ncbi.nlm.nih.gov/pubmed/31179301?tool=bestpractice.com
[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
[34]van Wassenaer-van Hall HN. Neuroimaging in Wilson disease. Metab Brain Dis. 1997 Mar;12(1):1-19.
http://www.ncbi.nlm.nih.gov/pubmed/9101534?tool=bestpractice.com
Hyperintensity on T2-weighted images of the basal ganglia
Atrophy of the head of the caudate nucleus, brainstem, and cerebral and cerebellar hemispheres.
The 'face of the giant panda' sign is seen in T2-weighted images of the midbrain. It is uncommon but, if present, is characteristic of Wilson's disease.[35]Gupta A, Chakravarthi S, Goyal MK. 'Face of giant panda': a rare imaging sign in Wilson's disease. QJM. 2014 Jul;107(7):579.
https://academic.oup.com/qjmed/article/107/7/579/1555920
http://www.ncbi.nlm.nih.gov/pubmed/24170891?tool=bestpractice.com
These findings are sensitive for Wilson's disease in cases of neurological presentation only. They are not specific for Wilson's disease. MRI findings can be similar in other neurological disorders.
DNA testing for mutations in the ATP7B gene
Although there are more than 700 pathogenic mutations in the ATP7B gene, next-generation sequencing can identify two pathogenic mutations in the majority of people with Wilson's disease.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
The absence of two pathogenic mutations does not exclude Wilson's disease; clinical corroboration is required.[24]Shribman S, Marjot T, Sharif A, et al; British Association for the Study of the Liver Rare Diseases Special Interest Group. Investigation and management of Wilson's disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022 Jun;7(6):560-75.
http://www.ncbi.nlm.nih.gov/pubmed/35429442?tool=bestpractice.com
Molecular testing is preferred before liver biopsy in children with suspected Wilson's disease.[25]Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-44.
https://journals.lww.com/jpgn/Fulltext/2018/02000/Wilson_s_Disease_in_Children__A_Position_Paper_by.32.aspx
http://www.ncbi.nlm.nih.gov/pubmed/29341979?tool=bestpractice.com
DNA analysis can be used to genotype siblings of a diagnosed Wilson's disease patient. It is not necessary to know the specific ATP7B mutation or mutations in the patient. DNA markers on both sides of both copies of the ATP7B gene in the patient are characterised. If a sibling matches both sets of markers they are affected, if they match one set they are a carrier, and if they match neither they are unlikely to have the disease.[19]Ala A, Borjigin J, Rochwarger A, et al. Wilson disease in septuagenarian siblings: raising the bar for diagnosis. Hepatology. 2005 Mar;41(3):668-70.
https://aasldpubs.onlinelibrary.wiley.com/doi/10.1002/hep.20601
http://www.ncbi.nlm.nih.gov/pubmed/15723329?tool=bestpractice.com