Progressive supranuclear palsy (PSP) is a clinical diagnosis based on the patient’s history and physical examination.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
PSP has multiple distinct phenotypic subtypes that vary in presentation. Oculomotor dysfunction, postural instability, akinesia, and cognitive dysfunction are the four characteristic clinical manifestations of typical PSP.
PSP is the most prevalent degenerative parkinsonian disorder after idiopathic Parkinson’s disease.[8]Swallow DMA, Zheng CS, Counsell CE. Systematic review of prevalence studies of progressive supranuclear palsy and corticobasal syndrome. Mov Disord Clin Pract. 2022 Jul;9(5):604-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9274340
http://www.ncbi.nlm.nih.gov/pubmed/35844273?tool=bestpractice.com
[29]Viscidi E, Litvan I, Dam T, et al. Clinical features of patients with progressive supranuclear palsy in an US insurance claims database. Front Neurol. 2021 Jun 17;12:571800.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8245849
http://www.ncbi.nlm.nih.gov/pubmed/34220661?tool=bestpractice.com
At present there are no laboratory biomarkers, imaging findings, or other investigations that can confirm a diagnosis of PSP during the patient’s lifetime. Definitive diagnosis requires postmortem pathology.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
The most prevalent subtype is the classic form of PSP, also known as PSP-Richardson’s syndrome (PSP-RS).[4]Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014 Dec;29(14):1758-66.
http://www.ncbi.nlm.nih.gov/pubmed/25370486?tool=bestpractice.com
[13]Morgan JC, Ye X, Mellor JA, et al. Disease course and treatment patterns in progressive supranuclear palsy: a real-world study. J Neurol Sci. 2021 Feb 15;421:117293.
http://www.ncbi.nlm.nih.gov/pubmed/33385754?tool=bestpractice.com
Other subtypes vary according to the early predominant symptoms/signs.
Consider the possibility of PSP if a patient aged 40 years or older presents with these typical features of classical PSP (PSP-RS):[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
Progressive falls (resulting from postural instability), especially within the first 3 years of the disease course and/or
Slow vertical eye movements (saccades)/vertical supranuclear gaze palsy. This typically causes the patient difficulty with tasks that require looking downwards (e.g., reading, eating).
Be aware that many patients with PSP are initially misdiagnosed with Parkinson's disease (PD) or other disorders that also present with parkinsonism, but a careful assessment for red flags points to the correct diagnosis in most patients.
As many as half of patients with PSP are initially diagnosed with PD.[29]Viscidi E, Litvan I, Dam T, et al. Clinical features of patients with progressive supranuclear palsy in an US insurance claims database. Front Neurol. 2021 Jun 17;12:571800.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8245849
http://www.ncbi.nlm.nih.gov/pubmed/34220661?tool=bestpractice.com
Examination of eye movements and postural stability, together with careful evaluation for other red flags, will point towards PSP as the correct diagnosis, particularly in the case of PSP-RS. A poor or absent response to levodopa is also an important clue to consider PSP. See History, below, for details.
In practice, the diagnosis of PSP is typically made 3-4 years after symptom onset, when the cardinal features of falls and supranuclear gaze palsy have become unequivocally apparent.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Clinical diagnosis of the non-Richardson’s syndrome PSP subtypes is challenging due to their considerable clinicopathological overlap with other conditions.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Other PSP phenotypes include PSP-parkinsonism (PSP-P, which mimics Parkinson’s disease), PSP with progressive gait freezing (PSP-PGF), PSP-corticobasal syndrome (PSP-CBS), PSP with speech/language disorders (PSP-SL), and PSP with frontal presentation (PSP-F).[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
If you suspect a patient may have any form of PSP, refer them to a movement disorder specialist for further evaluation as confirmation of the diagnosis requires specialist expertise.
History
PSP can present with a wide variety of symptoms, according to the distinct disease phenotype.
At least 8 different clinical phenotypes are known that can represent an underlying PSP pathology. Classic PSP (PSP-RS) and PSP-P are the most prevalent and the other phenotypes are rare.
Although clinical history and examination varies initially for different phenotypes, as the disease progresses all patients are likely to develop the core clinical features of PSP-RS (postural instability with falls and vertical oculomotor dysfunction).[4]Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014 Dec;29(14):1758-66.
http://www.ncbi.nlm.nih.gov/pubmed/25370486?tool=bestpractice.com
The presenting clinical phenotype for an individual patient will depend on the site of the most severe brain pathology and the pattern of its spread across synapses in the brain.
The presenting features for the main PSP phenotypes are outlined below.
Note that non-classic PSP phenotypes are hard to diagnose based on clinical findings. More commonly, they are diagnosed retrospectively based on autopsy findings.[14]Boxer AL, Yu JT, Golbe LI, et al. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017 Jul;16(7):552-63.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5802400
http://www.ncbi.nlm.nih.gov/pubmed/28653647?tool=bestpractice.com
PSP-Richardson’s syndrome (PSP-RS)
PSP-RS (also known as classic PSP) is the most common PSP presenting phenotype and is seen in about 40% to 50% of all people with a PSP pathology.[4]Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014 Dec;29(14):1758-66.
http://www.ncbi.nlm.nih.gov/pubmed/25370486?tool=bestpractice.com
[13]Morgan JC, Ye X, Mellor JA, et al. Disease course and treatment patterns in progressive supranuclear palsy: a real-world study. J Neurol Sci. 2021 Feb 15;421:117293.
http://www.ncbi.nlm.nih.gov/pubmed/33385754?tool=bestpractice.com
Symptoms usually start in the sixth or seventh decade of life and typically involve:[1]Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: a heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. 1964 Apr;10:333-59.
http://www.ncbi.nlm.nih.gov/pubmed/14107684?tool=bestpractice.com
[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
Postural instability leading to early unprovoked falls.
Characteristic abnormal eye movements: slowness of vertical saccades, progressing to vertical supranuclear gaze palsy, typically causing the patient difficulty with looking downwards. Slow vertical saccades are defined by a slow enough speed of vertical eye movements for the examiner to see the movements in a bedside test. Vertical supranuclear gaze palsy presents as a clear limitation of the range of voluntary gaze in the vertical more than the horizontal plane, affecting both upwards and downwards gaze.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Axial-predominant akinetic-rigid parkinsonism.
Speech and swallowing impairments.
Cognitive (mainly executive) dysfunction (e.g., difficulty planning/multitasking).
Be aware that many patients with PSP are initially incorrectly diagnosed with Parkinson’s disease or another parkinsonian disorder.[29]Viscidi E, Litvan I, Dam T, et al. Clinical features of patients with progressive supranuclear palsy in an US insurance claims database. Front Neurol. 2021 Jun 17;12:571800.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8245849
http://www.ncbi.nlm.nih.gov/pubmed/34220661?tool=bestpractice.com
Red flags that point towards PSP-RS as the correct diagnosis include:[30]McFarland NR, Hess CW. Recognizing atypical parkinsonisms: "red flags" and therapeutic approaches. Semin Neurol. 2017 Apr;37(2):215-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5961706
http://www.ncbi.nlm.nih.gov/pubmed/28511262?tool=bestpractice.com
[31]Bhidayasiri R, Sringean J, Reich SG, et al. Red flags phenotyping: a systematic review on clinical features in atypical parkinsonian disorders. Parkinsonism Relat Disord. 2019 Feb;59:82-92.
http://www.ncbi.nlm.nih.gov/pubmed/30409560?tool=bestpractice.com
[32]Williams DR, Litvan I. Parkinsonian syndromes. Continuum (Minneap Minn). 2013 Oct;19(5):1189-212.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4234134
http://www.ncbi.nlm.nih.gov/pubmed/24092286?tool=bestpractice.com
Postural instability/unsteady gait with falls (often backwards falls) in the first 3 years of disease onset[33]Brown FS, Rowe JB, Passamonti L, et al. Falls in progressive supranuclear palsy. Mov Disord Clin Pract. 2020 Jan;7(1):16-24.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6962663
http://www.ncbi.nlm.nih.gov/pubmed/31970205?tool=bestpractice.com
Slowness of vertical saccades or vertical supranuclear gaze palsy
Axial-predominant (trunk and neck) symmetric parkinsonism (bradykinesia, rigidity, and tremor) rather than asymmetric appendicular (limb) parkinsonism, with a lack of response to levodopa[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[34]European Reference Network for Rare Neurological Diseases (ERN-RND). Brief and focused information on the diagnosis of atypical parkinsonism for general neurologists/general practitioners. Jun 2020 [internet publication].
https://www.ern-rnd.eu/wp-content/uploads/2020/07/ERN-RND-General-information-for-general-neurologists-and-GPs_final.pdf
Early dysphagia and/or dysarthria
Significant early cognitive impairment mainly involving frontal executive function (e.g., difficulty planning/multitasking)
Freezing of gait not responding to levodopa
Rapid progression of symptoms.
Early postural instability occurring in the first 1-3 years of symptom onset is an important feature of PSP-RS. It presents with a sense of imbalance and repeated, usually backwards, falls.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
Neurodegeneration is rapidly progressive. Typically, within 3-4 years after symptom onset, patients become dependent on assistance for walking and the median survival has been reported as 6 to 7.4 years.[11]Bessemer R, Iansavichene A, Jenkins ME, et al. Clinical milestones as triggers for palliative care intervention in progressive supranuclear palsy and multiple system atrophy. J Neurol Sci. 2023 May 15;448:120614.
http://www.ncbi.nlm.nih.gov/pubmed/37001415?tool=bestpractice.com
[35]Golbe LI, Ohman-Strickland P, Beisser EB, et al. A convenient prognostic tool and staging system for progressive supranuclear palsy. Mov Disord Clin Pract. 2020 Aug;7(6):664-71.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7396865
http://www.ncbi.nlm.nih.gov/pubmed/32775512?tool=bestpractice.com
Ask about autonomic symptoms and check orthostatic blood pressure changes to distinguish PSP from multiple system atrophy (MSA). MSA is another parkinsonian disorder that is sometimes misdiagnosed as PSP because it also presents with early balance impairment and falls and typically has a poor response to levodopa. A key difference is that MSA is typically preceded by autonomic disturbances.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Slowing of vertical saccades, progressing to vertical supranuclear gaze palsy, is a defining feature of PSP-RS and usually presents within 2-3 years of symptom onset.[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
Ask about difficulty with tasks that require a downwards gaze, such as reading, eating, or descending stairs. Such tasks are often affected by the initial visual symptoms but the symptoms are not always spontaneously reported or questioned.
Some patients may complain of diplopia or blurred vision due to difficulty with convergence.
Speech impairment is an early symptom observed in half of PSP-RS patients.[36]Goetz CG, Leurgans S, Lang AE, et al. Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology. 2003 Mar 25;60(6):917-22.
http://www.ncbi.nlm.nih.gov/pubmed/12654953?tool=bestpractice.com
The patient's voice may be hoarse, growling, or slurred.
At advanced stages, the patient may lose the ability to speak (anarthria).
Impaired swallowing, which presents with frequent coughing on swallowing liquids and drooling (sialorrhoea), usually occurs within 1-2 years after speech disturbances.[36]Goetz CG, Leurgans S, Lang AE, et al. Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology. 2003 Mar 25;60(6):917-22.
http://www.ncbi.nlm.nih.gov/pubmed/12654953?tool=bestpractice.com
Mild cognitive impairment involving mainly the executive domain is frequent early in the disease course, leading to dementia in at least half of patients in more advanced stages.[37]Gerstenecker A, Mast B, Duff K, et al. Executive dysfunction is the primary cognitive impairment in progressive supranuclear palsy. Arch Clin Neuropsychol. 2013 Mar;28(2):104-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3569947
http://www.ncbi.nlm.nih.gov/pubmed/23127882?tool=bestpractice.com
[38]Pilotto A, Gazzina S, Benussi A, et al. Mild cognitive impairment and progression to dementia in progressive supranuclear palsy. Neurodegener Dis. 2017;17(6):286-91.
http://www.ncbi.nlm.nih.gov/pubmed/28881351?tool=bestpractice.com
Behavioural and cognitive symptoms are the presenting feature in around 20% of patients with PSP.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Patients usually complain of difficulties planning and multitasking. Memory disturbance is usually characterised by deficits in retrieval, with relatively preserved encoding (learning of new information).[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Significant memory impairment or hallucinations are rare in PSP and suggestive of other diagnoses.[39]Gerstenecker A, Duff K, Mast B, et al. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. 2013 Dec 30;210(3):1205-10.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3840159
http://www.ncbi.nlm.nih.gov/pubmed/24035530?tool=bestpractice.com
[40]Litvan I, Mega MS, Cummings JL, et al. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996 Nov;47(5):1184-9.
http://www.ncbi.nlm.nih.gov/pubmed/8909427?tool=bestpractice.com
Behavioural changes in the frontotemporal spectrum are commonly seen in patients with PSP.[39]Gerstenecker A, Duff K, Mast B, et al. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. 2013 Dec 30;210(3):1205-10.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3840159
http://www.ncbi.nlm.nih.gov/pubmed/24035530?tool=bestpractice.com
[40]Litvan I, Mega MS, Cummings JL, et al. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996 Nov;47(5):1184-9.
http://www.ncbi.nlm.nih.gov/pubmed/8909427?tool=bestpractice.com
Apathy and, less frequently, depression are the most common behavioural symptoms in PSP, presenting in about 60% of patients.[39]Gerstenecker A, Duff K, Mast B, et al. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. 2013 Dec 30;210(3):1205-10.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3840159
http://www.ncbi.nlm.nih.gov/pubmed/24035530?tool=bestpractice.com
[41]Flavell J, Nestor PJ. A systematic review of apathy and depression in progressive supranuclear palsy. J Geriatr Psychiatry Neurol. 2022 May;35(3):280-92.
http://www.ncbi.nlm.nih.gov/pubmed/33567955?tool=bestpractice.com
Impulsiveness and disinhibition are seen in one third of patients and can lead to behaviours that increase the risk of falls.[33]Brown FS, Rowe JB, Passamonti L, et al. Falls in progressive supranuclear palsy. Mov Disord Clin Pract. 2020 Jan;7(1):16-24.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6962663
http://www.ncbi.nlm.nih.gov/pubmed/31970205?tool=bestpractice.com
These features usually present as motor recklessness: for example, carelessness when standing and walking to pick up something off the floor despite severe balance/gait problems, or overstuffing the mouth despite impaired swallowing.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[39]Gerstenecker A, Duff K, Mast B, et al. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. 2013 Dec 30;210(3):1205-10.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3840159
http://www.ncbi.nlm.nih.gov/pubmed/24035530?tool=bestpractice.com
Pseudobulbar affect (PBA) may present in some patients and is characterised by labile emotions (e.g., inappropriate episodes of weeping or, less frequently, laughter). Anxiety is less frequent.[39]Gerstenecker A, Duff K, Mast B, et al. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. 2013 Dec 30;210(3):1205-10.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3840159
http://www.ncbi.nlm.nih.gov/pubmed/24035530?tool=bestpractice.com
[40]Litvan I, Mega MS, Cummings JL, et al. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996 Nov;47(5):1184-9.
http://www.ncbi.nlm.nih.gov/pubmed/8909427?tool=bestpractice.com
Other non-specific symptoms that are often reported by patients with PSP are:
Sleep disturbances. Disordered sleep is an under-recognised feature of PSP and is often progressive, leading to impaired daytime functioning.[42]Rowe JB, Holland N, Rittman T. Progressive supranuclear palsy: diagnosis and management. Pract Neurol. 2021 Oct;21(5):376-83.
https://pn.bmj.com/content/21/5/376.long
http://www.ncbi.nlm.nih.gov/pubmed/34215700?tool=bestpractice.com
Patients (or carers) might report insomnia, excessive daytime sleepiness, and sleep fragmentation (e.g., obstructive sleep apnoea, periodic limb movement disorder, and restless leg syndrome), although all of these are also common in other neurodegenerative disorders.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Constipation. This affects 71% to 80% of patients with PSP.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Putative causes include a sedentary lifestyle, hypodipsia, central dysautonomia, and medication adverse effects.
Urinary symptoms. These affect 53% to 93% of people with PSP and early onset indicates a worse prognosis.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Urgency, incontinence, and hesitancy are the most common urinary symptoms.[43]Reimann M, Schmidt C, Herting B, et al. Comprehensive autonomic assessment does not differentiate between Parkinson's disease, multiple system atrophy and progressive supranuclear palsy. J Neural Transm (Vienna). 2010 Jan;117(1):69-76.
http://www.ncbi.nlm.nih.gov/pubmed/19763772?tool=bestpractice.com
A urology referral is necessary to differentiate PSP-related symptoms from common age-related conditions such as benign prostatic hyperplasia in men and pelvic floor weakness in women.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
PSP-parkinsonism (PSP-P)
PSP-P is the second most common PSP phenotype, accounting for 30% to 40% of cases.[4]Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014 Dec;29(14):1758-66.
http://www.ncbi.nlm.nih.gov/pubmed/25370486?tool=bestpractice.com
[13]Morgan JC, Ye X, Mellor JA, et al. Disease course and treatment patterns in progressive supranuclear palsy: a real-world study. J Neurol Sci. 2021 Feb 15;421:117293.
http://www.ncbi.nlm.nih.gov/pubmed/33385754?tool=bestpractice.com
[44]Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord. 2010 Feb 15;25(3):357-62.
http://www.ncbi.nlm.nih.gov/pubmed/20108379?tool=bestpractice.com
Compared with classical PSP-RS, patients with a PSP-P phenotype have a more protracted disease course.
These patients present with asymmetric limb bradykinesia and rigidity that shows greater response to levodopa than other PSP subtypes. This is sometimes associated with rest tremor.
The defining PSP features of slow vertical saccades/supranuclear vertical gaze palsy and postural instability with falls typically appear later than in PSP-RS (usually ≥2 years after symptom onset).[44]Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord. 2010 Feb 15;25(3):357-62.
http://www.ncbi.nlm.nih.gov/pubmed/20108379?tool=bestpractice.com
Rarer PSP subtypes: PSP-frontal presentation (PSP-F)
PSP-F presents with cognitive dysfunction or behavioural changes due to frontal lobe involvement. Common signs and symptoms are executive dysfunction, apathy, perseveration, disinhibition, and impulsivity.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
[45]Han HJ, Kim H, Park JH, et al. Behavioral changes as the earliest clinical manifestation of progressive supranuclear palsy. J Clin Neurol. 2010 Sep;6(3):148-51.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2950920
http://www.ncbi.nlm.nih.gov/pubmed/20944816?tool=bestpractice.com
[46]Hassan A, Parisi JE, Josephs KA. Autopsy-proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase. 2012;18(6):478-88.
http://www.ncbi.nlm.nih.gov/pubmed/22181323?tool=bestpractice.com
Rarer PSP subtypes: PSP-speech/language disorder (PSP-SL)
PSP-SL is characterised by progressive apraxia of speech and non-fluent aphasia.[47]Josephs KA, Boeve BF, Duffy JR, et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase. 2005 Aug;11(4):283-96.
http://www.ncbi.nlm.nih.gov/pubmed/16093229?tool=bestpractice.com
Apraxia of speech is a speech disorder that affects motor function, resulting in slow and effortful speech, with abnormal rhythm, intonation, and emphasis of speech (dysprosody).
Non-fluent aphasia is defined by hesitant and non-fluent spontaneous speech, omission or impaired use of grammatical elements (agrammatism), and phonemic errors, while comprehension of word meaning remains intact.[48]Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol. 2008 Dec;21(6):688-92.
http://www.ncbi.nlm.nih.gov/pubmed/18989114?tool=bestpractice.com
[49]Santos-Santos MA, Mandelli ML, Binney RJ, et al. Features of patients with nonfluent/agrammatic primary progressive aphasia with underlying progressive supranuclear palsy pathology or corticobasal degeneration. JAMA Neurol. 2016 Jun 1;73(6):733-42.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4924620
http://www.ncbi.nlm.nih.gov/pubmed/27111692?tool=bestpractice.com
Note that speech apraxia and non-fluent aphasia are also associated with other neurodegenerative conditions, such as frontotemporal lobar degeneration and corticobasal degeneration.
Rarer PSP subtypes: PSP-corticobasal syndrome (PSP-CBS)
PSP-CBS is a rare phenotype that manifests with a combination of cortical and basal ganglia dysfunction symptoms. These include cortical sensory loss, ideomotor apraxia, alien limb phenomenon, limb dystonia, and asymmetric rigidity and akinesia that is commonly levodopa-resistant.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[50]Ling H, de Silva R, Massey LA, et al. Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol. 2014 Feb;40(2):149-63.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4260147
http://www.ncbi.nlm.nih.gov/pubmed/23432126?tool=bestpractice.com
Rarer PSP subtypes: PSP-progressive gait freezing (PSP-PGF)
PSP-PGF, which is very rare, presents with progressive freezing of gait (sudden and transient motor blocks or start hesitation when walking) within the first year of the disease, accompanied by bradykinesia and/or rigidity.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[51]Müller J, Seppi K, Stefanova N, et al. Freezing of gait in postmortem-confirmed atypical parkinsonism. Mov Disord. 2002 Sep;17(5):1041-5.
http://www.ncbi.nlm.nih.gov/pubmed/12360556?tool=bestpractice.com
[52]Rezvanian S, Litvan I, Standaert D, et al. Understanding the relationship between freezing of gait and other progressive supranuclear palsy features. Parkinsonism Relat Disord. 2020 Sep;78:56-60.
http://www.ncbi.nlm.nih.gov/pubmed/32731191?tool=bestpractice.com
Symptoms typically do not respond to levodopa and, after several years, patients may develop the typical features of PSP-RS.[53]Facheris MF, Maniak S, Scaravilli F, et al. Pure akinesia as initial presentation of PSP: a clinicopathological study. Parkinsonism Relat Disord. 2008 Aug;14(6):517-9.
http://www.ncbi.nlm.nih.gov/pubmed/18325816?tool=bestpractice.com
[54]Compta Y, Valldeoriola F, Tolosa E, et al. Long lasting pure freezing of gait preceding progressive supranuclear palsy: a clinicopathological study. Mov Disord. 2007 Oct 15;22(13):1954-8.
http://www.ncbi.nlm.nih.gov/pubmed/17724746?tool=bestpractice.com
Rarer PSP subtypes: Research-oriented intermediate phenotypes
PSP-oculomotor dysfunction (PSP-OM) is characterised by eye movement involvement (slowness of vertical saccades and vertical supranuclear gaze palsy) with mild or absent features of postural instability, akinesia, or cognitive impairment.[4]Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014 Dec;29(14):1758-66.
http://www.ncbi.nlm.nih.gov/pubmed/25370486?tool=bestpractice.com
[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
PSP-postural instability (PSP-PI) is characterised by early postural instability and delayed involvement of abnormal eye movements.[4]Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014 Dec;29(14):1758-66.
http://www.ncbi.nlm.nih.gov/pubmed/25370486?tool=bestpractice.com
These are intermediate phenotypes of PSP-RS that have been developed for early recognition of patients for inclusion in therapeutic research studies.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Risk factors
There is a paucity of research on risk factors that might predispose an individual to PSP.
The key risk factor is advancing age.[8]Swallow DMA, Zheng CS, Counsell CE. Systematic review of prevalence studies of progressive supranuclear palsy and corticobasal syndrome. Mov Disord Clin Pract. 2022 Jul;9(5):604-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9274340
http://www.ncbi.nlm.nih.gov/pubmed/35844273?tool=bestpractice.com
[9]Savica R, Grossardt BR, Bower JH, et al. Incidence and pathology of synucleinopathies and tauopathies related to parkinsonism. JAMA Neurol. 2013 Jul;70(7):859-66.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3707980
http://www.ncbi.nlm.nih.gov/pubmed/23689920?tool=bestpractice.com
The typical age of onset is the mid 60s.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
No autopsy-confirmed case has been demonstrated in an individual younger than age 40 years.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
In most patients, older age will be the only identifiable risk factor.
Random genetic mutations can also play a part.[16]Höglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011 Jun 19;43(7):699-705.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3125476
http://www.ncbi.nlm.nih.gov/pubmed/21685912?tool=bestpractice.com
[18]Chen JA, Chen Z, Won H, et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener. 2018 Aug 8;13(1):41.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6083608
http://www.ncbi.nlm.nih.gov/pubmed/30089514?tool=bestpractice.com
[19]Sanchez-Contreras MY, Kouri N, Cook CN, et al. Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol Neurodegener. 2018 Jul 9;13(1):37.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6038352
http://www.ncbi.nlm.nih.gov/pubmed/29986742?tool=bestpractice.com
Other reported risk factors are mainly environmental and include exposure to chromate/phosphate from textile dyeing and tanning or exposure to pesticides from well water.[20]Caparros-Lefebvre D, Golbe LI, Deramecourt V, et al. A geographical cluster of progressive supranuclear palsy in northern France. Neurology. 2015 Oct 13;85(15):1293-300.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4617163
http://www.ncbi.nlm.nih.gov/pubmed/26354981?tool=bestpractice.com
[21]Litvan I, Lees PS, Cunningham CR, et al. Environmental and occupational risk factors for progressive supranuclear palsy: case-control study. Mov Disord. 2016 May;31(5):644-52.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4861658
http://www.ncbi.nlm.nih.gov/pubmed/26854325?tool=bestpractice.com
A lower level of education, use of firearms, and stressful life events have also been associated with a higher risk of developing PSP, while use of oestrogen replacement therapy has been associated with a lower risk.[21]Litvan I, Lees PS, Cunningham CR, et al. Environmental and occupational risk factors for progressive supranuclear palsy: case-control study. Mov Disord. 2016 May;31(5):644-52.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4861658
http://www.ncbi.nlm.nih.gov/pubmed/26854325?tool=bestpractice.com
[22]Kelley KD, Peavy G, Edland S, et al. The role of stress as a risk factor for progressive supranuclear palsy. J Parkinsons Dis. 2017;7(2):377-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5952610
http://www.ncbi.nlm.nih.gov/pubmed/28409749?tool=bestpractice.com
[23]Kelley KD, Checkoway H, Hall DA, et al. Traumatic brain injury and firearm use and risk of progressive supranuclear palsy among veterans. Front Neurol. 2018 Jun 20;9:474.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6020251
http://www.ncbi.nlm.nih.gov/pubmed/29973911?tool=bestpractice.com
[24]Park HK, Ilango S, Charriez CM, et al. Lifetime exposure to estrogen and progressive supranuclear palsy: environmental and genetic PSP study. Mov Disord. 2018 Mar;33(3):468-72.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5840026
http://www.ncbi.nlm.nih.gov/pubmed/29460982?tool=bestpractice.com
Physical examination
Focus your physical examination on looking for the signs that are suggestive of the classical form of PSP (PSP-RS), PSP-P, or one of the rarer phenotypic subtypes of the condition.
Parkinsonism
Signs of parkinsonism include bradykinesia associated with rigidity, tremor, or postural instability.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Bradykinesia is examined by gait assessment and finger or toe tapping tests, which show significant slowing, and/or dampening of amplitude, compared with healthy individuals.
While both Parkinson’s disease (PD) and PSP exhibit similar parkinsonian signs, there are distinct differences that can point towards PSP as the more likely diagnosis.[30]McFarland NR, Hess CW. Recognizing atypical parkinsonisms: "red flags" and therapeutic approaches. Semin Neurol. 2017 Apr;37(2):215-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5961706
http://www.ncbi.nlm.nih.gov/pubmed/28511262?tool=bestpractice.com
[31]Bhidayasiri R, Sringean J, Reich SG, et al. Red flags phenotyping: a systematic review on clinical features in atypical parkinsonian disorders. Parkinsonism Relat Disord. 2019 Feb;59:82-92.
http://www.ncbi.nlm.nih.gov/pubmed/30409560?tool=bestpractice.com
[32]Williams DR, Litvan I. Parkinsonian syndromes. Continuum (Minneap Minn). 2013 Oct;19(5):1189-212.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4234134
http://www.ncbi.nlm.nih.gov/pubmed/24092286?tool=bestpractice.com
In most PSP phenotypes, bradykinesia and rigidity involve the axial skeleton (trunk and neck) more severely than the limbs compared with PD.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
The patient with PSP might report neck and back stiffness.
Tremor is less frequent in PSP than in Parkinson’s disease and, when present, is usually milder and bilateral.[42]Rowe JB, Holland N, Rittman T. Progressive supranuclear palsy: diagnosis and management. Pract Neurol. 2021 Oct;21(5):376-83.
https://pn.bmj.com/content/21/5/376.long
http://www.ncbi.nlm.nih.gov/pubmed/34215700?tool=bestpractice.com
[44]Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord. 2010 Feb 15;25(3):357-62.
http://www.ncbi.nlm.nih.gov/pubmed/20108379?tool=bestpractice.com
See Differentials.
Freezing of gait can be provoked by asking the patient to pass through a narrow space or to turn around while walking.
Postural instability
Patients with PSP-RS usually have an erect posture and tend to fall backwards due to imbalance.[55]Amano S, Skinner JW, Lee HK, et al. Discriminating features of gait performance in progressive supranuclear palsy. Parkinsonism Relat Disord. 2015 Aug;21(8):888-93.
http://www.ncbi.nlm.nih.gov/pubmed/26032992?tool=bestpractice.com
Imbalanced, slow, lurching walking on a wide base is reminiscent of a 'drunken sailor'.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
In contrast to the en bloc turning typical of PD, patients with PSP may pivot carelessly and retropulse, with a risk of falling backwards.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Evaluate postural instability with the pull test.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
This involves the examiner standing behind the patient and first gently pulling both shoulders, then doing so more vigorously a second time to disturb the patient’s balance.[55]Amano S, Skinner JW, Lee HK, et al. Discriminating features of gait performance in progressive supranuclear palsy. Parkinsonism Relat Disord. 2015 Aug;21(8):888-93.
http://www.ncbi.nlm.nih.gov/pubmed/26032992?tool=bestpractice.com
An abnormal pull test is confirmed if the patient needs to take more than two steps backwards to regain balance or if they need to be caught.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
A sit-to-stand test may reveal altered sequencing due to lack of trunk flexion and anterior weight shift.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Tendency to fall on pull testIankova V, et al; Movement Disorder Society-endorsed PSP Study Group. Parkinsonism Relat Disord 2020 Sep;78:200-3; used with permission
Oculomotor signs
The characteristic slowing of vertical saccades, which is a crucial feature for diagnosing PSP, is not an early sign, often taking 2-3 years to manifest.[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
Oculomotor dysfunction typically starts with frequent square wave jerks, progressing to slowing of vertical saccades and then to the cardinal sign of vertical supranuclear gaze palsy.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
This delay in the appearance of abnormal eye movements is a factor in delayed diagnosis of PSP. Moreover, early signs are subtle and might be missed in initial evaluations if not specifically tested.
A valuable sign that is not part of the formal diagnostic criteria but can be seen before restriction and slowing of saccades is a curvilinear path on downwards saccades (the 'round-the-houses' sign).[42]Rowe JB, Holland N, Rittman T. Progressive supranuclear palsy: diagnosis and management. Pract Neurol. 2021 Oct;21(5):376-83.
https://pn.bmj.com/content/21/5/376.long
http://www.ncbi.nlm.nih.gov/pubmed/34215700?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: The “round-the-houses” sign, which can often be seen before restriction and slowing of saccades. Note the lateral curvature of downward path of eye movement (yellow arrows)Rowe JB, et al. Practical Neurology 2021; 21: 376-83; used with permission [Citation ends].
To assess the saccades, instruct the patient to shift their gaze between two fixed visual points (such as the index finger and thumb) positioned on the right/left or top/bottom of their visual field.
First evaluate the horizontal saccades and then proceed to examine the vertical saccades. Normally, the trajectory of the eyeball is so fast that it can hardly be seen to be moving, whereas in PSP the speed of the eyeball movement is so slow that the trajectory can be easily seen.[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
In patients with PSP, this slowing is seen to affect vertical eyeball movements both earlier than, and to a greater degree than, horizontal eyeball movements.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
Reduction or loss of the optokinetic nystagmus (OKN), typically tested with OKN strips, confirms the loss of saccades.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Restriction of the vertical range (as opposed to speed) of voluntary eye movements occurs later, with low amplitude of the voluntary vertical gaze.
Use the doll’s head manoeuvre to activate the vertibulo-ocular reflex, thereby confirming the supranuclear nature of gaze palsy (i.e., involuntary vertical eye movements are preserved whereas voluntary eye movements are impaired).[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
This involves asking the patient to fix the eyes on a target while the head is passively moved up and down.
Slow vertical saccadesIankova V, et al; Movement Disorder Society-endorsed PSP Study Group. Parkinsonism Relat Disord 2020 Sep;78:200-3; used with permission
Ask the patient to fixate on a near target with eyes in the primary position. Macro square wave jerks can frequently be seen in patients with PSP and are often an early sign of oculomotor dysfunction.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Jerky involuntary movements of small amplitude will be seen that momentarily move the eyes horizontally away from the target, followed by returning to the target after an interval of less than a second.
These are not specific to PSP but tend to be more frequent, larger movements in PSP than in other conditions in which square wave jerks can occur (e.g., other parkinsonian conditions, Friedreich’s ataxia, Huntington’s chorea, and multiple sclerosis.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
[56]Otero-Millan J, Schneider R, Leigh RJ, et al. Saccades during attempted fixation in parkinsonian disorders and recessive ataxia: from microsaccades to square-wave jerks. PLoS One. 2013;8(3):e58535.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3596296
http://www.ncbi.nlm.nih.gov/pubmed/23516502?tool=bestpractice.com
[57]Shaffer DM, Krisky CM, Sweeney JA. Frequency and metrics of square-wave jerks: influences of task-demand characteristics. Invest Ophthalmol Vis Sci. 2003 Mar;44(3):1082-7.
https://iovs.arvojournals.org/article.aspx?articleid=2123904
http://www.ncbi.nlm.nih.gov/pubmed/12601033?tool=bestpractice.com
Square wave jerks can also occasionally be seen in healthy older adults.
Macro square wave jerksIankova V, et al; Movement Disorder Society-endorsed PSP Study Group. Parkinsonism Relat Disord 2020 Sep;78:200-3; used with permission
Cognitive dysfunction
Evaluate the patient’s cognitive function. Cognitive impairment frequently occurs in PSP and predominantly involves frontal executive function and verbal fluency.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
[37]Gerstenecker A, Mast B, Duff K, et al. Executive dysfunction is the primary cognitive impairment in progressive supranuclear palsy. Arch Clin Neuropsychol. 2013 Mar;28(2):104-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3569947
http://www.ncbi.nlm.nih.gov/pubmed/23127882?tool=bestpractice.com
[58]Burrell JR, Hodges JR, Rowe JB. Cognition in corticobasal syndrome and progressive supranuclear palsy: a review. Mov Disord. 2014 Apr 15;29(5):684-93.
http://www.ncbi.nlm.nih.gov/pubmed/24757116?tool=bestpractice.com
Use a screening scale to evaluate cognition. The Montreal Cognitive Assessment (MoCA) scale is preferred over the Mini Mental State Examination (MMSE) because it is more sensitive in detecting executive dysfunction.[59]Fiorenzato E, Weis L, Falup-Pecurariu C, et al. Montreal Cognitive Assessment (MoCA) and Mini-Mental State Examination (MMSE) performance in progressive supranuclear palsy and multiple system atrophy. J Neural Transm (Vienna). 2016 Dec;123(12):1435-42.
http://www.ncbi.nlm.nih.gov/pubmed/27334897?tool=bestpractice.com
MoCA test: digital tools
Opens in new window
MoCA test: paper versions
Opens in new window
Speech/language impairment
Free speech will reveal most speech and/or language deficits. Language disturbances in PSP are characterised by:[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[36]Goetz CG, Leurgans S, Lang AE, et al. Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology. 2003 Mar 25;60(6):917-22.
http://www.ncbi.nlm.nih.gov/pubmed/12654953?tool=bestpractice.com
[60]Peterson KA, Patterson K, Rowe JB. Language impairment in progressive supranuclear palsy and corticobasal syndrome. J Neurol. 2021 Mar;268(3):796-809.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7914167
http://www.ncbi.nlm.nih.gov/pubmed/31321513?tool=bestpractice.com
Apraxia of speech (difficulty planning and coordinating the movements necessary for speech production, resulting in effortful, halting speech with inconsistent sound errors and distortions, or slow speech with syllables segmented)
Non-fluent aphasia (reduced fluency and difficulty in generating speech and forming coherent sentences).
Apraxia of speechIankova V, et al; Movement Disorder Society-endorsed PSP Study Group. Parkinsonism Relat Disord 2020 Sep;78:200-3; used with permission
nfa PPA spontaneous speechIankova V, et al; Movement Disorder Society-endorsed PSP Study Group. Parkinsonism Relat Disord 2020 Sep;78:200-3; used with permission
Bulbar signs
Dysarthria eventually affects almost all patients with PSP and has a hypokinetic and/or spastic feature, often accompanied by a strained-strangled or breathy voice quality.[61]Rusz J, Bonnet C, Klempíř J, et al. Speech disorders reflect differing pathophysiology in Parkinson's disease, progressive supranuclear palsy and multiple system atrophy. J Neurol. 2015;262(4):992-1001.
http://www.ncbi.nlm.nih.gov/pubmed/25683763?tool=bestpractice.com
[62]Clark HM, Utianski RL, Ali F, et al. Motor speech disorders and communication limitations in progressive supranuclear palsy. Am J Speech Lang Pathol. 2021 Jun 18;30(3s):1361-72.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8702836
http://www.ncbi.nlm.nih.gov/pubmed/33719524?tool=bestpractice.com
Swallowing impairment is frequent in PSP.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Choking or coughing on liquids and later on solid food are indicative symptoms, along with drooling.
If not apparent from the history, swallowing function can be evaluated by a water swallow test.
Pseudobulbar affect (PBA), sometimes classified as a behavioural symptom, affects many patients with PSP.
PBA is a representation of bulbar upper motor neuron involvement and is usually accompanied by a spastic dysarthria.[63]Verny M, Jellinger KA, Hauw JJ, et al. Progressive supranuclear palsy: a clinicopathological study of 21 cases. Acta Neuropathol. 1996;91(4):427-31.
http://www.ncbi.nlm.nih.gov/pubmed/8928621?tool=bestpractice.com
It presents with labile facial expression of emotions that might not be congruent with the patient’s mood.
Dystonia
Limb dystonia is usually unilateral, involving an upper limb or one side of the body.
It occurs most frequently in the corticobasal syndrome variant of PSP (PSP-CBS) but is also seen in up to one third of PSP-RS cases.[64]Barclay CL, Lang AE. Dystonia in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1997 Apr;62(4):352-6.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1074090
http://www.ncbi.nlm.nih.gov/pubmed/9120447?tool=bestpractice.com
Neck dystonia, although less frequent, is more specific to PSP (mainly PSP-RS).[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Facial dystonia can sometimes present in patients with PSP, resulting in deep nasolabial folds and a furrowed brow, giving the appearance of surprise or concern (referred to as the Procerus sign).[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[65]Golbe LI, Davis PH, Lepore FE. Eyelid movement abnormalities in progressive supranuclear palsy. Mov Disord. 1989;4(4):297-302.
http://www.ncbi.nlm.nih.gov/pubmed/2811889?tool=bestpractice.com
Disturbances in eyelid motion are frequent, including severely decreased blinking, lid retraction (resulting in a staring appearance), blepharospasm, and eyelid-opening apraxia. Blepharospasm and eyelid-opening apraxia both result in involuntary eyelid closure. Blepharospasm is caused by active contraction in the orbicularis oculi muscles, which may be amenable to treatment with botulinum toxin, whereas apraxia of eyelid opening is caused by an inability to open the eye due to central mechanisms and is far more difficult to treat.
Eyelid opening apraxiaIankova V, et al; Movement Disorder Society-endorsed PSP Study Group. Parkinsonism Relat Disord 2020 Sep;78:200-3; used with permission
Other signs
Neurological examination will reveal apraxia in many patients with PSP.[66]Leiguarda RC, Pramstaller PP, Merello M, et al. Apraxia in Parkinson's disease, progressive supranuclear palsy, multiple system atrophy and neuroleptic-induced parkinsonism. Brain. 1997 Jan;120 (Pt 1):75-90.
https://academic.oup.com/brain/article/120/1/75/312814
http://www.ncbi.nlm.nih.gov/pubmed/9055799?tool=bestpractice.com
Apraxia is defined as a disorder of skilled action and tool use despite adequate sensorimotor and cognitive function. It is one of the defining features in the PSP-CBS phenotype, but is also frequently seen in PSP-RS (if the degree of motor impairment allows for appropriate testing).[67]Soliveri P, Piacentini S, Girotti F. Limb apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology. 2005 Feb 8;64(3):448-53.
http://www.ncbi.nlm.nih.gov/pubmed/15699373?tool=bestpractice.com
Bedside tests include asking the patient to copy gestures (e.g., a peace sign or OK sign) or asking the patient to mime how they would use a tool to perform a task (e.g., brushing teeth or stirring coffee).
Myoclonus is an important feature of the CBS phenotype that rarely occurs in PSP-RS.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Pyramidal signs such as brisk muscle stretch reflexes, Babinski sign, Hoffmann sign, or exaggerated jaw jerk are occasionally present in patients with various phenotypes of PSP.[68]Stejskalova Z, Rohan Z, Rusina R, et al. Pyramidal system involvement in progressive supranuclear palsy - a clinicopathological correlation. BMC Neurol. 2019 Mar 20;19(1):42.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6425568
http://www.ncbi.nlm.nih.gov/pubmed/30894142?tool=bestpractice.com
Initial investigations
Imaging
Request a brain magnetic resonance imaging (MRI) in any case where PSP is suspected.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[69]American College of Radiology. ACR appropriateness criteria: movement disorders and neurodegenerative diseases. 2019 [internet publication].
https://acsearch.acr.org/docs/3111293/Narrative
Although PSP is diagnosed clinically, an initial brain MRI helps to rule out other differential diagnoses such as normal pressure hydrocephalus, midbrain or third ventricular tumour, pineal gland tumour, frontal mass lesions, multiple small vessel infarcts, and vascular parkinsonism.
Demonstration of the characteristic features of PSP can increase diagnostic confidence in a patient who has been diagnosed based on clinical features, meaning the patient qualifies for an 'imaging supported diagnosis'.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
MRI in PSP is characterised by significant mesencephalic atrophy. Atrophy of midbrain and superior cerebellar peduncles strongly supports PSP.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[14]Boxer AL, Yu JT, Golbe LI, et al. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017 Jul;16(7):552-63.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5802400
http://www.ncbi.nlm.nih.gov/pubmed/28653647?tool=bestpractice.com
[70]Albrecht F, Bisenius S, Neumann J, et al. Atrophy in midbrain and cerebral/cerebellar pedunculi is characteristic for progressive supranuclear palsy: a double-validation whole-brain meta-analysis. Neuroimage Clin. 2019;22:101722.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6402426
http://www.ncbi.nlm.nih.gov/pubmed/30831462?tool=bestpractice.com
This feature can result in various radiographic signs, including 'hummingbird', 'Mickey Mouse', and 'morning glory' signs in the midsagittal and axial planes.[14]Boxer AL, Yu JT, Golbe LI, et al. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017 Jul;16(7):552-63.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5802400
http://www.ncbi.nlm.nih.gov/pubmed/28653647?tool=bestpractice.com
While this pattern is very frequent in PSP-RS, it is unclear how early this feature appears. Moreover, it is not usually present in the other phenotypes; therefore, its absence does not exclude PSP.
The Magnetic Resonance Parkinsonism Index (MRPI) - calculated as the pons-midbrain width ratio multiplied by the middle cerebellar peduncles-superior cerebellar peduncles width ratio - has a high sensitivity and specificity for distinguishing PSP from Parkinson’s disease (PD).[71]Nigro S, Morelli M, Arabia G, et al. Magnetic Resonance Parkinsonism Index and midbrain to pons ratio: which index better distinguishes progressive supranuclear palsy patients with a low degree of diagnostic certainty from patients with Parkinson disease? Parkinsonism Relat Disord. 2017 Aug;41:31-6.
http://www.ncbi.nlm.nih.gov/pubmed/28487107?tool=bestpractice.com
[72]Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008 Jan;246(1):214-21.
http://www.ncbi.nlm.nih.gov/pubmed/17991785?tool=bestpractice.com
[73]Kim S, Suh CH, Shim WH, et al. Diagnostic performance of the Magnetic Resonance Parkinsonism Index in differentiating progressive supranuclear palsy from Parkinson’s disease: an updated systematic review and meta-analysis. Diagnostics (Basel). 2021 Dec 22;12(1):12.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8774886
http://www.ncbi.nlm.nih.gov/pubmed/35054178?tool=bestpractice.com
A newer version, MRPI 2.0, has high sensitivity and specificity (100% and 94.3%, respectively) in differentiating the parkinsonism subtype of PSP (PSP-P) from PD.[74]Quattrone A, Morelli M, Nigro S, et al. A new MR imaging index for differentiation of progressive supranuclear palsy-parkinsonism from Parkinson's disease. Parkinsonism Relat Disord. 2018 Sep;54:3-8.
http://www.ncbi.nlm.nih.gov/pubmed/30068492?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: MRI of the brain in a patient with clinically diagnosed PSP. Image A: sagittal T1-MRI through the brainstem demonstrates atrophy in the midbrain with relative preservation of the pons, giving the appearance of a hummingbird. Image B: on axial T1-weighted imaging, the dorsal midbrain is reduced in volume, giving a “Mickey Mouse” appearanceSchott JM. Practical Neurology 2007; 7: 186-190; used with permission [Citation ends].
The European Association of Nuclear Medicine/European Academy of Neurology and the American College of Radiology recommend fluorodeoxyglucose positron emission tomography (FDG-PET) imaging as potentially helpful in distinguishing PSP (in particular, the PSP-RS phenotype) from PD, particularly in the first 2 years of symptom onset.[69]American College of Radiology. ACR appropriateness criteria: movement disorders and neurodegenerative diseases. 2019 [internet publication].
https://acsearch.acr.org/docs/3111293/Narrative
[75]Nobili F, Arbizu J, Bouwman F, et al. European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain (18) F-fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: Delphi consensus. Eur J Neurol. 2018 Oct;25(10):1201-17.
https://onlinelibrary.wiley.com/doi/10.1111/ene.13728
http://www.ncbi.nlm.nih.gov/pubmed/29932266?tool=bestpractice.com
In PSP-RS, hypometabolism is typically seen, whereas patients with PD generally show normal or even increased striatal metabolism.[75]Nobili F, Arbizu J, Bouwman F, et al. European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain (18) F-fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: Delphi consensus. Eur J Neurol. 2018 Oct;25(10):1201-17.
https://onlinelibrary.wiley.com/doi/10.1111/ene.13728
http://www.ncbi.nlm.nih.gov/pubmed/29932266?tool=bestpractice.com
Therapeutic trial
A trial of levodopa (in the form carbidopa/levodopa) is strongly recommended for patients who are affected by parkinsonism. Minimal or no response supports a diagnosis of PSP.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Progressively increase the dose to the recommended range.
Continue the highest tolerated dosage for at least 1 month before determining the response.
Levodopa resistance sufficient to support a diagnosis of PSP is defined as ≤30% improvement on the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS).[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
The parkinsonism subtype of PSP (PSP-P) may be an exception where levodopa will elicit a more robust response.
Other investigations
Other investigations can be ordered based on clinical clues to rule out differential diagnoses that might mimic PSP.
A tilt table test can identify neurogenic orthostatic hypotension, which would point towards a diagnosis of multiple system atrophy (MSA) or Lewy body dementia rather than PSP.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
In practice, consider performing an active stand test first, with measurement of blood pressure (BP) and heart rate in the supine and standing position. A significant drop in BP with no significant increase in heart rate from supine to standing is indicative of neurogenic orthostatic hypotension.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
A tilt table test is indicated if the active stand test is inconclusive.
Consider requesting polysomnography (where available) if a patient with parkinsonism has a clear history of dream enactment, obtained from a bed partner. It may occasionally be helpful in distinguishing PSP from PD, dementia with Lewy bodies (DLB), and MSA.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Rapid eye movement (REM) sleep behaviour disorder is uncommon in PSP and more supportive of a diagnosis of PD, DLB, or MSA. However, be aware that its presence does not exclude PSP, with reported prevalence in this patient group ranging from 14% to 33%.[10]Bluett B, Pantelyat AY, Litvan I, et al. Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. Front Neurol. 2021 Jul 1;12:694872.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284317
http://www.ncbi.nlm.nih.gov/pubmed/34276544?tool=bestpractice.com
Several treatable disorders can present with vertical supranuclear gaze palsy. Specific diagnostic tests can help to differentiate these disorders from PSP. These include:
For Neimann-Pick disease type C (NPC): bone marrow biopsy or skin biopsy with fibroblast culture and filipin test
For Whipple's disease: polymerase chain reaction (PCR) for Tropheryma whippelii on a cerebrospinal fluid (CSF) sample or intestinal biopsy[76]Magherini A, Pentore R, Grandi M, et al. Progressive supranuclear gaze palsy without parkinsonism: a case of neuro-Whipple. Parkinsonism Relat Disord. 2007 Oct;13(7):449-52.
http://www.ncbi.nlm.nih.gov/pubmed/17071126?tool=bestpractice.com
For Wilson’s disease: a 24-hour urine copper may suggest the diagnosis, warranting further investigation
For autoimmune/paraneoplastic encephalitis: serum and CSF paraneoplastic antibody panel.
If disease progression is particularly rapid, CSF biomarkers to exclude prion disease may be indicated.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Emerging investigations
Biomarkers are being developed that can help differentiate PSP from other neurodegenerative parkinsonian disorders including corticobasal degeneration, frontotemporal lobar degeneration, multiple system atrophy, Lewy body dementia, and even Alzheimer’s disease.[14]Boxer AL, Yu JT, Golbe LI, et al. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017 Jul;16(7):552-63.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5802400
http://www.ncbi.nlm.nih.gov/pubmed/28653647?tool=bestpractice.com
For example, if Alzheimer’s disease is suspected, beta-amyloid or tau PET scan can help rule out this disorder.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
However, even where available these biomarkers are not routinely used in clinical practice because an exact pathological diagnosis does not change the clinical management in most cases due to the lack of specific disease-modifying treatments.
Certainty of diagnosis
The formal diagnostic criteria for PSP are complex and categorise the patient as having probable, possible, or suggestive PSP according to a range of factors. A definitive diagnosis requires postmortem neuropathological examination.
The 2017 Movement Disorder Society (MDS) diagnostic criteria for the full spectrum of PSP phenotypic subtypes cover basic features that must be present, a wide range of exclusion criteria that rule out PSP, clinical and imaging clues that can support a PSP diagnosis, and a table of core clinical features for different symptom domains (ocular motor dysfunction/postural instability/akinesia/cognitive dysfunction). The criteria are used to determine the level of likelihood that a patient has PSP (probable/possible/suggestive) and the phenotypic subtype.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
Prior to publication of the 2017 MDS criteria, the 1996 criteria published by the National Institute of Neurological Disorders and Stroke/Society for PSP (NINDS/SPSP criteria) were used.[28]Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.
http://www.ncbi.nlm.nih.gov/pubmed/8710059?tool=bestpractice.com
These have excellent specificity but limited sensitivity for non-Richardson's syndrome PSP subtypes.[3]Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord. 2017 Jun;32(6):853-64.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516529
http://www.ncbi.nlm.nih.gov/pubmed/28467028?tool=bestpractice.com
See Criteria.
 Tendency to fall on pull test
Tendency to fall on pull test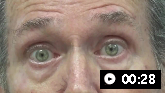 Slow vertical saccades
Slow vertical saccades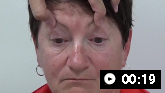 Vertical supranuclear gaze palsy with demonstration of doll’s head manoeuvre
Vertical supranuclear gaze palsy with demonstration of doll’s head manoeuvre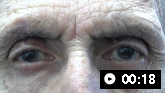 Macro square wave jerks
Macro square wave jerks Apraxia of speech
Apraxia of speech nfa PPA spontaneous speech
nfa PPA spontaneous speech Eyelid opening apraxia
Eyelid opening apraxia