Article Text

Abstract
Background Neoadjuvant treatment has become standard for patients with high-risk operable stage III melanoma, but the optimal regimen is unknown. Targeted therapy approaches yield high pathological response rates, while immunotherapy regimens show favorable recurrence-free survival (RFS). NeoACTIVATE was designed to address whether a neoadjuvant combination of both targeted therapy and immunotherapy might leverage the benefits of each.
Methods We tested neoadjuvant treatment with 12 weeks of vemurafenib, cobimetinib, and atezolizumab for patients with BRAF-mutated (BRAFm) melanoma (cohort A) and cobimetinib and atezolizumab for patients with BRAF-wild-type (BRAFwt) melanoma (cohort B), regimens which we have shown generate a substantial major pathological response. After therapeutic lymph node dissection, patients received 24 weeks of adjuvant atezolizumab. Here, we report survival outcomes and their association with biomarkers assayed among the gut microbiome and peripheral blood immune subsets.
Results With 49 months median follow-up, the median RFS was not reached for cohort A and was 40.8 months for cohort B. At 24 months after operation, 2 of 14 cohort A patients and 4 of 13 cohort B patients had experienced distant relapse. Key findings from correlative analyses included diversity, taxonomic and functional metagenomic gut microbiome signals associated with distant metastasis-free survival at 2 years. Notably, we observed a strong correlation between low microbial arginine biosynthesis (required for T-cell activation and effector function) and early distant recurrence (p=0.0005), which correlated with taxonomic differential abundance findings. Peripheral blood immune monitoring revealed increased double-positive (CD4+CD8+) T cells in patients with early recurrence.
Conclusions Neoadjuvant treatment with cobimetinib and atezolizumab±vemurafenib was associated with a low rate of distant metastasis in patients with high-risk stage III melanoma. Freedom from early distant metastasis was highly associated with taxonomic differences in gut microbiome structure and with functional pathway alterations known to modulate T cell immunity. Identification of predictive biomarkers will permit optimization of neoadjuvant therapy regimens for individual patients.
Trial registration number NCT03554083.
- Skin Cancer
- Surgery
- Immune Checkpoint Inhibitor
- Immunotherapy
- Neoadjuvant
Data availability statement
Data are available on reasonable request. Immediately following publication, individual participant data that underlie the results reported in this article, after deidentification (text, tables, figures, and appendices) will be shared on request with researchers who provide a methodologically sound proposal for analyses to achieve the aims in the approved proposal. Raw microbiome sequence data will be deposited into SRA (https://www.ncbi.nlm.nih.gov/sra). On request, we will provide deidentified data with scientific approval of the study team under restricted access via email request. These data will be provided to scientific investigators for research purposes. Requests will be reviewed by the Institutional Review Board and subject to a Data Use Agreement. Details on acceptable methods and duration of data transfer will be determined by institutional policies based on which data are requested and for what research purposes. Proposals should be directed to hieken.tina@mayo.edu.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
When this study opened, little data existed to support the use of neoadjuvant therapy for operable high-risk stage III melanoma, but now several trials support the clinical benefit of neoadjuvant immunotherapy for these patients. The impact of combining targeted and immunotherapy on survival outcomes is not well understood.
WHAT THIS STUDY ADDS
Here, we show a clinically meaningful benefit in survival outcomes from neoadjuvant combinatorial therapy. With over 4 years median follow-up, longer than in similar studies, we observed a very low rate of distant metastatic disease in these high-risk patients. Moreover, we demonstrate that the composition of the gut microbiome, both in terms of microbial taxa and functional pathways, is highly associated with distant metastasis within 2 years.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our study adds to the growing body of evidence supporting neoadjuvant treatment prior to operation for patients with high-risk but operable stage III melanoma. While the majority of trials have shown favorable outcomes using immune checkpoint inhibitor combinations, ours demonstrates benefit from the combination of immunotherapy and targeted therapy and suggests potential biomarkers of response that merit further investigation.
Introduction
Neoadjuvant therapy has been associated with favorable outcomes and is rapidly being accepted as the preferred treatment for surgically resectable clinically detected stage III melanoma.1–6 Although early modern neoadjuvant trials of targeted therapy regimens showed a high pathological response rate in patients with BRAF-mutated (BRAFm) patients, concern arose that these did not translate into durable remissions, particularly once treatment was discontinued.7 8 This raises the question of whether a short course of targeted therapy combined with concurrent immunotherapy might lead to more durable disease control while driving a higher pathological response rate to induction therapy.
While mitogen-activated protein kinase (MAPK) pathway-targeted therapies have not proven beneficial for BRAF-wild-type (BRAFwt) melanomas, chronic tumor-specific T cell receptor (TCR) signaling through MAPK kinase (MEK) drives T cell exhaustion. Transient inhibition of MEK minimizes T cell exhaustion and supports more robust antitumor immune responses. Moreover, the combination of transient MEK inhibition with targeting of the PD-1/PD-L1 axis has been demonstrated to be synergistic in preclinical tumor models.9
Even with the most aggressive neoadjuvant treatment regimens, considerable variability in both pathological response and freedom from melanoma recurrence exists among treated patients. The availability of an increasing number of options for neoadjuvant treatment, coupled with the heterogeneity in response to a given regimen, mandates the identification of biomarkers of treatment response that would permit selection of the most efficacious and least toxic neoadjuvant approach for each patient.
We designed a two-cohort phase II clinical trial (NeoACTIVATE, NCT03554083) testing the combination of targeted and immunotherapy for patients with clinically detected stage III melanoma. Patients were assigned treatment by BRAF mutation status, with BRAFm patients (cohort A) treated with neoadjuvant vemurafenib (BRAF inhibitor), cobimetinib (MEK inhibitor), and atezolizumab (PD-L1 blocking antibody); and BRAFwt patients (cohort B) treated with neoadjuvant cobimetinib and atezolizumab. Patients then underwent restaging, therapeutic lymph node dissection, and adjuvant therapy with atezolizumab. Tissue, blood, and microbiome specimens were collected prior to and after neoadjuvant treatment. We have previously reported out on the pathological response endpoint of NeoACTIVATE cohorts A and B and observed major pathological responses (≤10% viable tumor) in 2/3 of BRAF-mutated patients and 1/3 of BRAF-wild-type patients.10 Here, we report on clinical outcomes following adjuvant treatment, including recurrence-free survival (RFS), distant metastasis-free survival (DMFS), adverse events, and key translational studies in association with these outcome measures.
Methods
Study design and participants
NeoACTIVATE (NCT03554083) is an investigator-initiated, open label, two-cohort, phase II multicenter clinical trial carried out at Mayo Clinic (Rochester, Minnesota, USA) and the University of Minnesota (Minneapolis, Minnesota, USA), whose primary adjuvant aim is to examine RFS among patients with high-risk, resectable stage III BRAF-mutant melanoma receiving vemurafenib-cobimetinib-atezolizumab (cohort A) and among BRAF-wild-type patients receiving cobimetinib-atezolizumab (cohort B). The primary aim of the neoadjuvant portion of NeoACTIVATE—pathological response to neoadjuvant treatment—was previously reported.10 Full details of the protocol, inclusion and exclusion criteria are provided in the protocol (available as online supplemental file 2 published with that manuscript). Enrollment to NeoACTIVATE was previously described.10
Supplemental material
Interventions and assessments
All patients had pretreatment needle biopsy performed. Neoadjuvant and adjuvant treatment were given according to online supplemental table 1. Patients were restaged with cross-sectional imaging after cycle 3 and then proceeded to operation (TLND). Adverse events during neoadjuvant and adjuvant therapy were assessed using the CTCAE V.5.0 criteria. Patients were followed by clinical examination and cross-sectional imaging every 12 weeks while on adjuvant therapy and for 2.5 years thereafter or until recurrence.
Supplemental material
Formalin-fixed, paraffin-embedded tissue was obtained for research purposes both at the time of preregistration biopsy and at the time of TLND. Blood was obtained at the time of registration, after cycles 1, 2, and 3 of neoadjuvant therapy, and after cycle 4 (TLND); blood samples were processed to viably cryopreserve peripheral blood mononuclear cells (PBMCs) and to preserve plasma per laboratory protocols. Stool samples from baseline and after cycle 3 of neoadjuvant treatment were collected via collection kits supplied to the patient, frozen by the patient, and transferred to the primary research laboratory. Buccal and skin swabs were obtained prior to cycle 1 treatment and after cycle 3. Plasma and microbiome specimens were stored at −80°C for batched analysis, while PBMCs were stored under liquid nitrogen. The number of research biospecimens obtained for each timepoint is listed in online supplemental table 2.
PD-L1 immunohistochemistry
PD-L1 immunohistochemistry was performed centrally and evaluated by the study pathologist (TJF) as previously described.10 Briefly, tissue slides were stained using the clone 22C3 monoclonal antibody (Agilent Dako, Santa Clara, California, USA), and scoring was performed independently for tumor cells and immune cells as assessed semiquantitatively.
Microbiome assessments and analysis
DNA extraction was performed using 250 mg of each sample using the Dneasy PowerSoil Pro Kit (Qiagen, USA) following the manufacturer’s protocol. After quality control analysis and library prep (Illumina, San Diego, USA), shallow shotgun metagenomic sequencing was performed (CosmosID Inc, Germantown, Maryland, USA) and yielded 3931 unique Amplicon Sequence Variants. The average total read count across all samples was 2.64492×106 (median 2.52–8895×106; range 8.5563×105 to 9.829×106). Taxonomic profiling was performed using Sourmash V.4.2.4 using the “sourmash gather” and “sourmash taxonomy” commands with GTDB202 used as a reference database.11 Functional profiling of metagenomic data was performed using HUMAnN V.3.6 with the Struo2 release of GTDB V.202 formatted for HUMAnN used as a reference.12 13 Gene families identified by HUMAnN were mapped to MetaCyc pathways and normalized to copies-per-million using the utility scripts provided with HUMAnN.
α-diversity and β-diversity were analyzed for species-level abundance data after rarefaction. α-diversity (within-sample diversity) reflects species richness and evenness within the microbial populations. Three α-diversity indices were calculated on the rarefied species data: observed number of species, Shannon index, and Inverse Simpson index. In contrast, β-diversity (between-sample diversity) reflects the shared diversity between bacterial populations in terms of ecological distance; pair-wise distance measure allows quantification of the overall compositional difference between samples. Different β-diversity measures provide distinctive views of the community structure. Multiple β-diversity measures: unweighted, generalized (α=0.5) and weighted UniFrac distances, Jaccard distance, and Bray-Curtis distance were calculated using the species abundance table and the phylogenetic tree.14
To test the association between clinical covariates and α-diversity measures, a linear regression-based t-test was used. To test the association between the covariates and β-diversity measures, we used PERMANOVA, a distance-based analysis of variance method based on permutation.15 Ordination plots were generated using principal coordinate analysis for visualizing the association of covariates with the β-diversities. Taxonomic differential abundance analysis was performed using the LinDA method, a linear model based on centered log ratio-transformed data that addresses compositional effects in microbiome data.16 Species with a prevalence less than 10% or with a maximum proportion less than 0.2% were excluded from testing to reduce the number of tests. Functional differential abundance analysis was performed using MaAsLin2.17 Minimum feature abundance was set to 2×10–4; all other parameters were set to default values. False discovery rate (FDR) control via the Benjamini-Hochberg (B-H) procedure18 was used to correct for multiple testing, and FDR-adjusted p values or q values <0.1 were considered significant.
Quantitation of sPD-L1 in plasma
Soluble PD-L1 was measured by ELISA as previously described.10 ELISAs were performed by technologists who were blinded to the identity of the samples.
Mass cytometry of peripheral immune cells
Mass cytometry was performed on PBMCs using a panel of antibodies and techniques10 as previously described. For analysis, sample FCS files were uploaded to the Astrolabe Cytometry Platform (Astrolabe Diagnostics) and subjected to automated transformation, quality control, and unsupervised clustering. Processed cellular count and marker expression were exported and further analyzed using R (V.4.3.1). To assess cellular population abundance, cellular counts were normalized as a percentage of identified PBMCs per subject at each timepoint. Significant differences between time points were determined for paired samples within each cohort using Wilcoxon’s signed-rank test, with significance defined as p<0.05. For grouped comparisons by recurrence status at each timepoint, a linear model t-test was fitted adjusting cohort membership, and significance was defined as p<0.1 after applying B-H FDR control.
Flow cytometry of T cell subsets and activation states
Flow cytometry was performed on PBMCs from baseline, after cycle 1, after cycle 3, and after surgery using an antibody panel designed to identify tumor-related T cells, effector cytotoxic T lymphocytes (CTLs), and pro-apoptotic T cells as previously described.10
Statistical considerations
Pathological responses and toxicities from neoadjuvant treatment within each cohort were previously reported.10 For results assessed after the start of adjuvant therapy, all patients who began adjuvant treatment were included in the analysis. RFS, distant RFS (DRFS), and adverse events (emerging or worsening during/after adjuvant treatment) were assessed. RFS is defined as the time from surgery to radiographic or histological evidence of local, regional, or distant recurrence of melanoma or death due to any cause. For DMFS, some patients had regional recurrence before distant recurrence, which may affect distant recurrence. So a competing risk approach was used to assess DRFS. Cumulative incidence estimates of DMFS were computed using the Fine and Gray method,19 where regional recurrence was considered a competing event. RFS at 2 years (RFS-2) is defined as no regional recurrence, distant recurrence, or death 2 years after TLND. DMFS at 2 years (DMFS-2) is defined as no distant recurrence or death within 2 years. New and worsening adverse events (CTCAE V.5, deemed at least possibly related to treatment) during adjuvant treatment were analyzed in a tabular method. Statistical analyses were performed using SAS software, V.9.4. Data were frozen as of March 4, 2024.
Results
Clinical
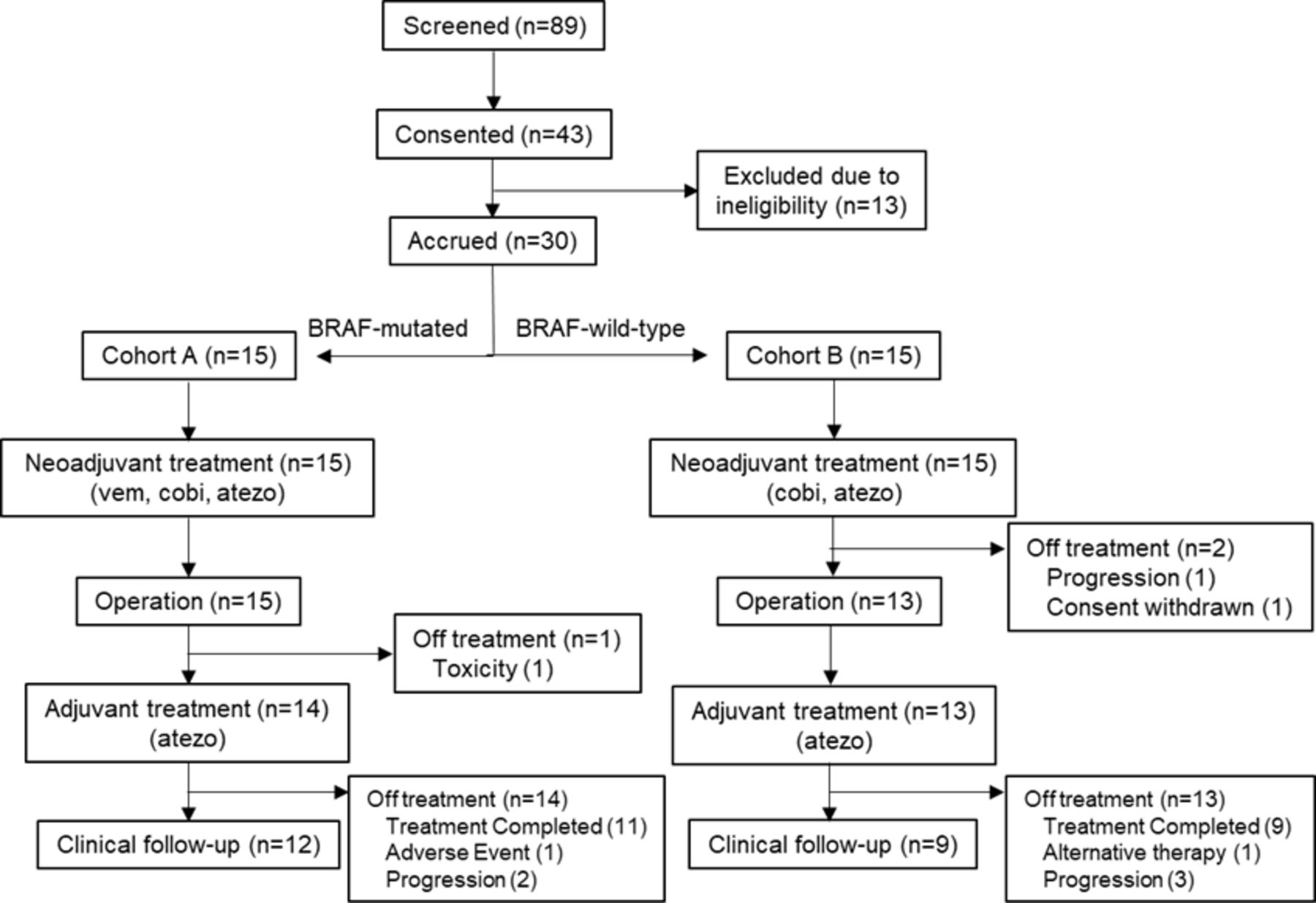
Between June 22, 2018 and May 10, 2021, patients were enrolled in cohorts A (n=15) and B (n=15) of NeoACTIVATE. After completion of 12 weeks of neoadjuvant therapy, 28 patients (15 cohort A, 13 cohort B) proceeded to a per-protocol operation. One cohort A patient experienced grade 3 pneumonitis and went off protocol within 1 month of the operation to pursue alternative (targeted) treatment. Therefore, 27 patients (14 cohort A, 13 cohort B) initiated adjuvant treatment with atezolizumab and were evaluable for RFS, cumulative incidence of distant metastasis (CIDM), and safety outcomes of adjuvant therapy (figure 1). Patient characteristics are summarized in table 1.

CONSORT diagram. Study participation outcomes for all screened patients are shown. CONSORT, Consolidated Standards of Reporting Trials.
Demographic and tumor characteristics of patients operated on and initiating adjuvant therapy on protocol
Of the 14 patients in cohort A, 3 received fewer than 8 adjuvant cycles: 1 due to adverse events (grade 2 encephalitis, received 3 adjuvant cycles), 1 due to disease progression (received 4 adjuvant cycles), and 1 due to patient refusal (persistent grade 1 arthralgias, received 6 adjuvant cycles). Of the 13 patients in cohort B, 4 did not complete 8 adjuvant cycles: 3 due to disease progression (after receiving 7, 4, and 2 adjuvant cycles, respectively) and 1 to pursue alternative therapy (after receiving 1 adjuvant cycle). No grade 3 or higher adverse events were observed during or after adjuvant therapy. No continuing adverse events worsened (higher grade) from neoadjuvant to adjuvant treatment. The most common new grade 2 adverse events emerging during adjuvant therapy were arthralgia (two cohort A and two cohort B patients) and maculopapular rash (three cohort B patients), as summarized in online supplemental table 3.
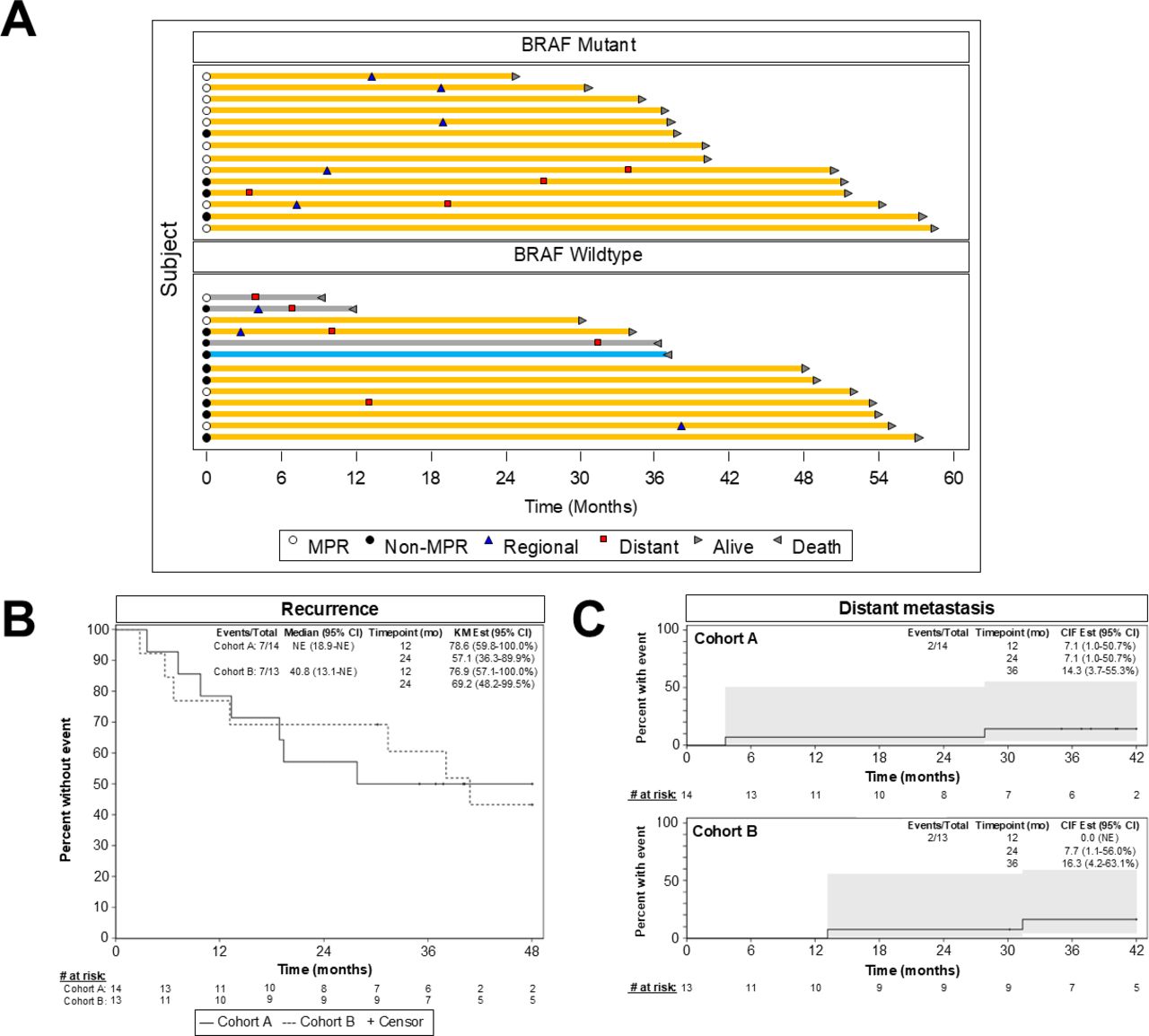
All patients have been followed for a minimum of 24 months from the date of operation (range 24–58.4 months, median follow-up 49 months for surviving patients). 13 (7 cohort A, 6 cohort B) are alive without recurrence, 4 (3 cohort A, 1 cohort B) are alive following a regional recurrence, 6 (4 cohort A, 2 cohort B) are alive with distant recurrence (with or without preceding or concomitant regional recurrence), and 4 (all cohort B) have died (1 with no recurrence—death unrelated to melanoma or treatment, 3 with distant recurrence). Timing of regional and distant recurrences is shown in the Swimmer plot, figure 2A.

Survival outcomes. (A) Follow-up times, regional (blue triangle) and distant (red square) recurrence events, and mortality events (reverse arrowhead) for all subjects who completed a per-protocol operation and initiated adjuvant therapy. Yellow bars represent patients alive at data cut-off, gray bars represent patients who died from melanoma, and the blue bar represents a patient who died of an unrelated cause (sepsis) without melanoma recurrence. Open and closed circles denote patients with versus without a major pathological response (MPR) (≤10% viable tumor), respectively. (B) Kaplan-Meier curves were used to depict recurrence-free survival for each cohort. (C) The cumulative incidence of distant metastasis for each cohort.
Calculated from the date of operation, median RFS was not reached for cohort A and 40.8 months for cohort B, with 57.1% (95% CI 36.3% to 89.9%) of cohort A patients and 69.2% (95% CI 48.2% to 99.5%) of cohort B patients alive and recurrence-free at 24 months (figure 2B). Of 10 cohort A patients with a pCR or near-pCR, 5 were alive and recurrence-free at 24 months, whereas 5 had experienced recurrence; of 5 cohort B patients with a pCR or near-pCR, 4 were alive and recurrence-free at 24 months, whereas 1 had recurred (online supplemental table 4.) CIDMs at 24 months was 7.1% for cohort A and 7.7% for cohort B patients, as detailed in figure 2C.
Gut microbiome structure
We performed shallow shotgun metagenomic sequencing of stool samples at baseline and postneoadjuvant timepoints and correlated these findings with pathological response, early relapse (RFS-2), and early distant metastasis (DMFS-2). While there was no correlation between microbiome features and pathological response, and only weak associations with RFS-2, our analyses demonstrated significant associations with early distant metastasis (DMFS-2) (figure 3). We observed a trend toward lower α-diversity (within-sample species richness and evenness) at both time points in patients who developed distant metastasis within 2 years of operation. Similarly, multiple β-diversity measures exhibited statistically significant shifts in overall microbial community structure by DMFS-2 status at both timepoints. Differentially abundant taxa included 6 genera and 21 species level differences at baseline and 22 genera and 20 species after neoadjuvant therapy prior to operation. Baseline differences in patients who developed vs did not develop distant metastasis within 2 years included increased relative abundance of species of Anaerostipe, Bifidobacterium, Blautia, Collinsella, Lawsonibacter, Meriplasma, Pelethousia, and Sellimonas and a significant decrease in the genera Clostridiales and Faecalibacterium and species of Alistipes, Bariatricus, and Eisenbergiella. Across both time points, nine microbes consistently correlated with distant metastasis status: Blautia A caecimuris, Blautia sp00432195, Collinsella aeorfasciens Y, Collinsella aeorfasciens J, Collinsella sp900541065, Lawsonibacter sp00177015, Meterraneibacter norwichensis, and Merdiplasma sp905207905 were uniformly enriched, whereas Alistipes onderdonkii was depleted in patients who developed early distant metastasis.

Gut microbiome structure in association with early distant metastasis. (A) The phylum-level abundance profiles across subjects for patients with and without early distant metastasis. (B, C) Differences in α-diversity and β-diversity, respectively, by multiple measures. (D) Volcano plots identify the most significant differentially abundant taxa (x-axis: effect size in log2 fold change, y-axis: −log FDR-adjusted p value). The heatmap in (E) depicts the relative abundance of nine taxa differentially abundant between those with and without early distant metastasis at both time points. FDR, false discovery rate.
Gut microbiome functional pathways
Functional metabolic pathway assessment of baseline samples was correlated with pathological response, RFS-2, and DMFS-2. While no clear correlations were identified with pathological response or RFS-2, we found 20 pathways which differed significantly between patients with and without early distant metastasis (figure 4A). Two key metabolic pathways were markedly diminished in patients with early distant metastasis, including the microbial metabolic pathway of arginine biosynthesis (p=0.0005) and the pathway of unsaturated fatty acid biosynthesis, which was totally absent (p=0.01) among patients who developed early distant disease (figure 4B). Analysis of the relationship between microbial species and functional pathways showed inverse correlations between Blautia A wexlerae and the arginine biosynthesis pathway and between Gemmiger qucibialis and the unsaturated fatty acid biosynthesis pathway.

Gut microbiome functional pathways in association with early distant metastasis. (A) Volcano plot illustrates the most significantly differentially abundant pathways based on MaAsLin2. (B) Boxplots detail the relative expression of two key metabolic pathways with highly significant differences between patient groups.
Immune cell profiling
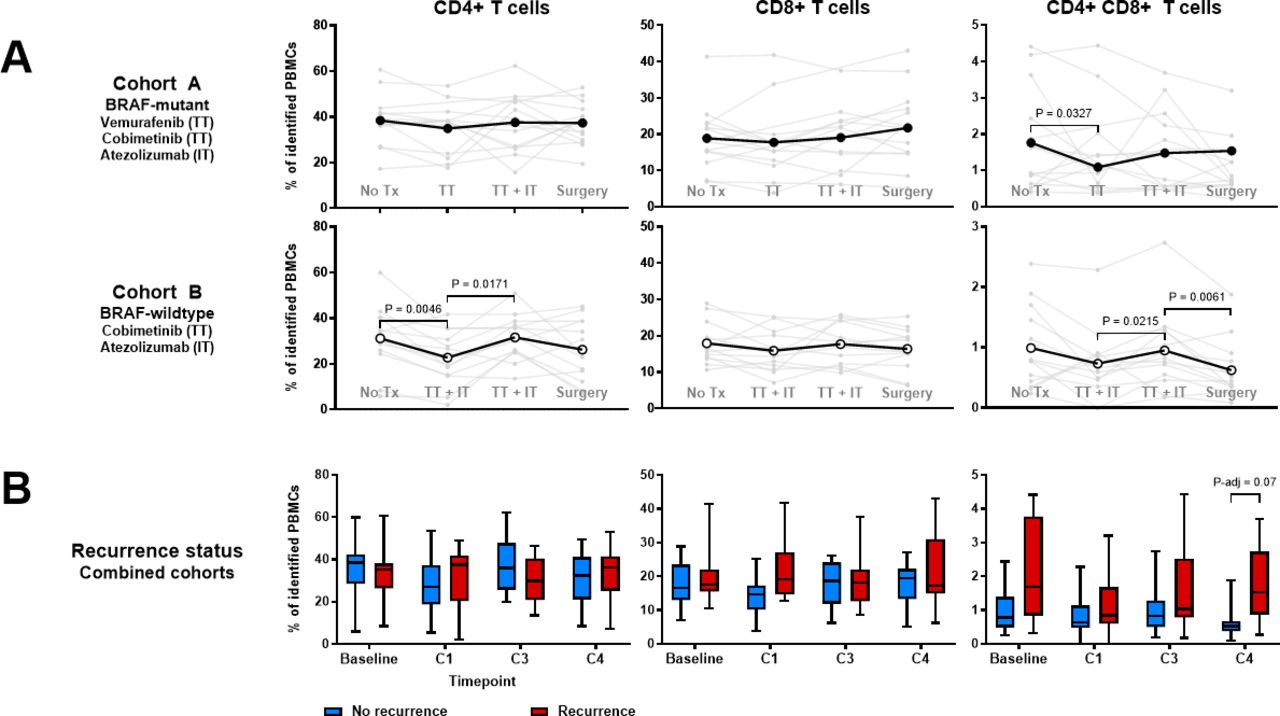
We employed mass cytometry to assess peripheral immune cell population changes in relation to treatment and outcomes using a comprehensive panel of immune cell lineage markers and relevant costimulatory and coinhibitory molecules.10 Initiation of neoadjuvant treatment was associated with transient decreases in CD4+T cells in cohort B and CD4+CD8+ double-positive (DP) T cells in both cohorts (figure 5A). DP T cells remained decreased after operation among patients in cohort B, while DP T cells remained elevated among patients in both cohorts who experienced recurrence (figure 5B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment-related T cell population changes. Time points indicated are baseline (prior to treatment), C1 (following one cycle of neoadjuvant therapy), C3 (following completion of neoadjuvant therapy), and C4 (after operation, prior to adjuvant therapy). (A) Individual and mean proportions of peripheral blood CD4+T cells, CD8+T cells, and CD4+CD8+ (DP) T cells as quantitated via mass cytometry by treatment cohort. Significance was defined as p<0.05 as measured by Wilcoxon’s signed rank test. (B) T cell populations by absence or presence of recurrence within 2 years, with median (middle line), IQR (box), and minimum and maximum (whiskers) indicated. Significance was determined using a linear model T-test, adjusting for treatment cohort and applying FDR correction. FDR, false discovery rate.
For greater resolution, we additionally used flow cytometry to quantitate distinct peripheral blood CD8+ T cell subpopulations, including tumor-related T cells, effector CTLs, and pro-apoptotic T cells. Both the baseline frequencies of each of these cell populations and the change in frequencies from before to after neoadjuvant treatment were highly variable and not obviously different between patients with vs without distant metastasis (online supplemental figure 1).
Tissue and soluble PD-L1
We correlated baseline and post-neoadjuvant tissue PD-L1 expression by immunohistochemistry with RFS. No significant associations between RFS and tissue PD-L1 were observed. These data are summarized in online supplemental figure 2. Plasma soluble PD-L1 (sPD-L1) concentrations varied widely between patients. Both post-neoadjuvant concentrations of sPD-L1 and the fold change in sPD-L1 from before to after neoadjuvant treatment were similar between patients with versus without distant metastasis within 2 years of operation (online supplemental figure 3).
Discussion
Here, we show that a short course of the neoadjuvant combination of targeted therapy and immune checkpoint blockade leads to favorable recurrence-free and DMFS in a substantial portion of patients with high-risk stage III melanoma. To the best of our knowledge, this is the first neoadjuvant study to test the combination of a targeted agent plus immune checkpoint blockade in patients with BRAFwt melanoma. For patients with BRAFm melanoma, neoadjuvant targeted therapy trials have yielded pCR rates of approximately 49%–58%.7 8 These compare favorably with pCR rates to single-agent anti-PD-1 (21%–25%)1 4 and are comparable to pCR rates from dual-agent immunotherapy trials (45%–57%).4 5 20 However, the durability of remission has been a concern with pure targeted therapy approaches, with a reported 5-year RFS of 40% after neoadjuvant/adjuvant dabrafenib/trametinib21 vs 70% after ipilimumab/nivolumab.3 Interestingly, while the 5-year outcomes from the NeoCombi trial21 suggested that patients exclusively had recurrence within the first 3 years, we observed one delayed (>3 years) melanoma recurrence in our patient population.
Similar to cohort A of NeoACTIVATE, the recently published NeoTrio trial tested combined targeted and immunotherapy in the neoadjuvant setting for patients with clinically detected stage III BRAFm melanoma.22 In contrast to NeoACTIVATE, NeoTrio had three arms to which BRAFm melanoma patients were randomized: immunotherapy alone (pembrolizumab), targeted therapy followed by immunotherapy (dabrafenib and trametinib for 7 days, then pembrolizumab), and concurrent targeted and immunotherapy (dabrafenib, trametinib, and pembrolizumab). Additional differences between the two studies were baseline extent of disease (29% N1b, 7% N2b, and 64% N3b in NeoACTIVATE vs 63% N1b, 20% N2b, and 17% N3b in NeoTrio), duration of neoadjuvant therapy (12 weeks in NeoACTIVATE vs 6 weeks in NeoTrio), and duration of follow-up (49 months in NeoACTIVATE vs 24.5 months in NeoTrio). Both studies showed a relatively high rate of pathological complete or near-complete response in patients treated with combined targeted therapy and immunotherapy.
A pooled analysis of multiple neoadjuvant therapy trials in stage III melanoma patients demonstrated that patients achieving a pCR, near-pCR, or pPR after neoadjuvant immunotherapy each had >90% 2-year RFS; whereas patients achieving a pCR after neoadjuvant targeted therapy had a 79% 2-year RFS.23 In light of this dichotomy, we correlated 2-year RFS with pathological response in our patients who received combined targeted and immunotherapy. We found that, as with patients treated with purely targeted therapy, patients achieving a pCR after vemurafenib, cobimetinib, and atezolizumab were still at considerable risk for recurrence. Similarly, patients treated with concurrent immunotherapy and targeted therapy in the NeoTrio trial were also at risk for recurrence even if they achieved a pCR or near-pCR.23 These data suggest that pathological response is less predictive of survival outcomes when targeted therapy is combined with immunotherapy in the neoadjuvant setting vs a pure immunotherapy regimen. However, given the higher pathological response rates seen with combined neoadjuvant therapy, the higher frequency of recurrences in patients achieving a pathological complete/near complete response does not necessarily imply lower overall efficacy. Instead, in order to compare the efficacy of combined therapy versus pure immunotherapy, survival outcomes rather than pathological response rates alone must be compared. Given the favorably low rate of early distant metastasis we observed, combined targeted and immunotherapy for BRAFm patients may be a viable alternative to pure immunotherapy approaches in some patients, particularly if we can identify a biomarker predictive of non-response to neoadjuvant immunotherapy alone. Whether or not targeted therapy might be of greater benefit if continued in the adjuvant phase, with or without concurrent immunotherapy, remains unknown. Given the toxicity observed in the neoadjuvant phase of arm A of our study, the duration of adjuvant combinatorial treatment might need to be truncated to less than the standard of 12 months of systemic therapy.
To the best of our knowledge, ours is also the first study to explore microbial associations with the development of distant metastatic disease in high-risk stage III melanoma patients treated with neoadjuvant immunotherapy, rather than RFS in general (which may include surgically curable regional relapse). We found striking differences in (1) beta diversity (between sample community structure), (2) relative abundance of multiple taxa, and (3) relevant functional pathways. Most prior studies on the impact of the gut microbiome on immunotherapy response have evaluated patients with already established distant metastatic disease,24–30 with just two studies addressing stage III melanoma patients treated with neoadjuvant immunotherapy,31 32 both focusing on microbial associations with pathological response and one on stage III melanoma patients treated with adjuvant therapy only.33 Here, we report on multiple features of the microbiome that distinguish stage III patients with early distant relapse after treatment with neoadjuvant combinatorial therapy. It is not clear why strong associations were seen only between microbial features and early distant metastasis and not with pathological response or early recurrence, but it may be explained by the impact of targeted therapy on pathological response in this study and limits of discovery within a small sample set.
In our study, the relative abundance of several fecal microbes distinguished patients with early distant relapse. We observed enrichment at both time points of Blautia A caecimuris in patients with early distant recurrence, previously reported to correlate positively with CD8+T cell infiltration in colon cancer and with advancing breast cancer stage.34 35 While Blautia species are generally regarded as beneficent gut commensals, associated with anti-inflammatory effects via suppression of NF-kB, data are mixed, or contextual, with at least one species, Blautia schinkii inversely associated with RFS in the stage III melanoma adjuvant therapy report.33 Another microbe we found enriched in patients with early distant relapse, Sellimonas intestinalis, was reported to increase in relative abundance over time in patients with advanced melanoma with progression within 1 year, as well as to correlate positively with CD8+T cell infiltration in the aforementioned colon cancer study and with aggressive breast in a different study.30 34 36 Both are members of the Lachnospiraceae family, which was observed to be enriched in patients without an MPR to neoadjuvant therapy by Davar et al.32 Significantly depleted taxa included Clostridiales and Faecalibacterium at the genus level, and Allistipes onderdonkii and Gemmiger qucibaialis. Simpson et al reported a positive correlation with pathological response to neoadjuvant PD-1 and CTLA-4 blockade and relative abundance of Faecalibacterium prausnitzii as well as decreased relative abundance of the order Clostridiales in the stool microbiome of patients with both a poor response to and immune-related adverse events,31 associations also reported in several prior studies in advanced melanoma.24–26 28 Allistipes onderdonkii was associated with a favorable response to anti-PD-1 immunotherapy in lung cancer patients and to both anti-PD-1 and anti-PD-L1 therapy in biliary tract cancers,37 38 while Gemmiger species were associated with improved response to anti-CTLA-4 treatment in stage IV melanoma.24 These depleted microbes all produce short-chain fatty acids which may in turn regulate host immunity by influencing T cell differentiation and effector function.39–41
We also saw differences in key immune response-related microbial metabolic pathways between patients with versus without distant metastasis within 2 years, including a strong correlation between early distant metastasis and low arginine biosynthesis (required for T cell activation) as well as the total absence of the unsaturated fatty acid biosynthesis pathway. Arginine, known to enhance T cell proliferation and effector function, is often depleted in the tumor microenvironment to facilitate tumor immune evasion, and low baseline plasma arginine levels are predictive of poor treatment response in cancer patients treated with immune checkpoint inhibitors.42 A group has also used engineered probiotic microbes to convert ammonia to L-arginine, producing increased tumor-infiltrating lymphocytes and synergizing with anti-PD-L1 therapy when evaluated in preclinical tumor models.43 Arginine degradation blockade via inhibition of its catalytic enzymes ARG1 and/or ARG2 also is being explored as a strategy to enhance the response to immunotherapy.42 44 Thus, the evaluation of patient microbiome arginine utilization may help inform the utility of these therapeutic candidates. Unsaturated fatty acids have well-described anti-inflammatory effects and are important modulators of immune signaling pathways, from suppression of NF-κB to direct antiproliferative effects in cancer cells.45 Enrichment of the pathway for biosynthesis of unsaturated fatty acids was associated with improved overall survival and chemotherapy sensitivity in colon cancer, and thus should also be considered as a candidate predictor of immunotherapy response in future studies.46
In assessing PBMCs, we identified a postsurgical elevation in DP T cells among patients who experience recurrence. It has previously been shown that DP T cells are enriched intratumorally and can recognize melanoma cells in an antigen-specific manner while in the tumor.47 In peripheral blood, DP T cells have been described in patients with urological malignancies, where they have been shown to favor a T helper 2 (Th2)-like phenotype that would prototypically impair an antitumor immune response.48 Given that our analysis focused on peripheral blood immune cells, we hypothesize that this DP T cell population within our study may reflect Th2-like cells, comporting with the observation that patients who experience recurrence possess relatively high percentages of DP T cells after operation. To be sure, we did not directly test whether treatment augments the Th2-like phenotype of this subset. Furthermore, recent studies have identified elevated DP T cells within tumors as a marker of successful response to immune checkpoint blockade; future studies focused on lineage tracking may disambiguate the phenotypic differences between peripheral and intratumoral DP T cells and may definitively establish whether treatment impacts their function.49
Limitations of this study are those inherent to a small phase II study, including limited power to detect correlations and associations. Given the number of subjects in each cohort of the study, there was insufficient statistical power to perform sex-based or gender-based analyses. Strengths of this work include a median follow-up of 49 months, with complete 2-year follow-up of all patients, permitting us to report on DMFS.
Taken together, our clinical and translational findings support a clinically meaningful benefit of this neoadjuvant combinatorial approach, with very low rates of early distant relapse. These data suggest hypotheses to test regarding predictive and prognostic biomarkers. With now several potential neoadjuvant strategies for high-risk stage III melanoma patients, further investigation is planned to identify actionable strategies to help select the optimal neoadjuvant approach for individual patients.
Supplemental material
Data availability statement
Data are available on reasonable request. Immediately following publication, individual participant data that underlie the results reported in this article, after deidentification (text, tables, figures, and appendices) will be shared on request with researchers who provide a methodologically sound proposal for analyses to achieve the aims in the approved proposal. Raw microbiome sequence data will be deposited into SRA (https://www.ncbi.nlm.nih.gov/sra). On request, we will provide deidentified data with scientific approval of the study team under restricted access via email request. These data will be provided to scientific investigators for research purposes. Requests will be reviewed by the Institutional Review Board and subject to a Data Use Agreement. Details on acceptable methods and duration of data transfer will be determined by institutional policies based on which data are requested and for what research purposes. Proposals should be directed to hieken.tina@mayo.edu.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Mayo Clinic Institutional Review BoardIRB# 17-009383. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank Kevin Pavelko (Mayo Clinic Immune Monitoring Core), Alisha Birgin, Renee Bradshaw, Heidi Turner, Simone Veum, Lynn Flickinger, Jill Schimke, and Vicki Taylor for assistance with generation of mass cytometry data, development and conduct of the NeoACTIVATE clinical trial, and preparation of the manuscript.
References
Footnotes
X @DrMaraPiltin
Contributors Conceptualization: MSB and TJH; Methodology and analyses: MSB, GDN, SJ, JC, TJF, EPG, LAK, LY, CLE, CAS, HD, DZ and TJH; Trial coordination: MSB, ED-M and TJH; Investigation: MSB, SJ, JC, TJF, EPG, RRM, LAK, LY, ED-M, SNM, AD, HNM, MAP, DLP, SSK, JYCH, CLE and TJH; Writing: MSB, GDN, SJ, JC, LY, EPG, CAS, HD and TJH; Final editorial review: all authors; Supervision: MSB and TJH; Funding: MSB and TJH. MSB and TJH are the joint guarantors.
Funding This study was funded by a Stand Up to Cancer-Genentech Catalyst Research Grant Award SU2C-AACR-CT1017, and Mayo Clinic Center for Clinical and Translational Science (cCaTS) Award 92541640-2020/Mayo Clinic. The indicated Stand Up To Cancer research grant is administered by the American Association for Cancer Research, the scientific partner of SU2C.
Disclaimer Neither Genentech nor Stand Up To Cancer was involved in the writing of the manuscript or the decision to submit it for publication. The authors have not been paid to write this article by a pharmaceutical company or other agency. The authors were not precluded from accessing data in the study, and they accept responsibility to submit for publication.
Competing interests MSB: Research support—Alkermes, Bristol-Myers Squibb, Genentech, Merck, nFerence, Perspective Therapeutics, Pharmacyclics, Regeneron, Sorrento, TILT Biotherapeutics, and Transgene; Consultant/Scientific Advisory Board—Perspective Therapeutics, Sorrento Therapeutics, TILT Biotherapeutics; RRM: Research support—Glaxo SmithKline and Bristol-Myers Squibb; LAK: Consultant/Scientific Advisory Board—Immunocore; SNM: Research support—Bristol-Myers Squibb; Royalties—Sorrento Therapeutics and Journey Therapeutics; AD: Research support—Syntrix Pharmaceuticals, Novartis, Merck, AnHearth Therapeutics, Sorrento Therapeutics, Guardant, Philogen, AstraZeneca; Consultant/Scientific Advisory Board—TP Therapeutics, Guardant Health, AnHeart Therapeutics, Chromacode; Honoraria—Intellisphere; MAP: Research support—Intuitive Surgical; DLP: Consultant/Scientific Advisory Board—In Situ Biologics; TJH: Research support—Genentech and SkylineDX BV.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.