Article Text

Abstract
Background Lynch syndrome (LS), caused by germline pathogenic variants in the mismatch repair genes, leads to high rates of frameshift-derived neopeptide (FSDN) expression due to microsatellite instability (MSI). While colorectal cancer (CRC) prevention is effective, most LS-related tumors lack such strategies. Cancer vaccines targeting FSDNs offer a promising approach for immune interception in LS. This study aimed to identify and validate LS-related FSDNs to develop vaccines for cancer prevention.
Methods We identified LS-related coding MS mutations and predicted FSDN with high coverage on common Human Leukocyte Antigen (HLA)-I and II alleles. We validated FSDN-associated mutations in colorectal adenomas (CrAD), endometrial cancers (EC), and CRC samples from patients with LS, non-LS tumors, and cell lines. Immunogenicity was assessed through interferon (IFN)-γ enzyme-linked immunospot and flow cytometry analysis of tissue-infiltrating lymphocytes (TILs) from LS carriers.
Results We prioritized 53 HLA-I and 45 HLA-II FSDNs in MSI tumors using in silico predictions. Validation revealed 86.7% of FSDN-associated mutations present in LS-CRC samples, with a median of 7.67 (6.5–9) mutations in CrADs and 6.02 (2–10) in CRCs. Sequencing of CrAD and EC samples showed 95% and 77.5% of predicted FSDN-associated mutations, respectively. MSI cancer cell lines transcribed 69.8% of FSDNs. Immunogenicity assays showed that 71% of potential FSDNs elicited IFN-γ responses, with a median of 7.37 (1–10.75) HLA-I and 6 (2–5.75) HLA-II FSDNs per patient. After prioritizing 24 FSDN, in a cohort of 19 LS-derived samples (4 CrAD and 15 normal mucosa), 52% (10/19) demonstrated T-cell reactivity to an HLA-I neoantigen pool. CD8+CD137+ activation markers increased significantly (p=0.037) over time and peptide-specific cells were detected by pentamer staining.
Conclusions Our predicted FSDN set has optimal coverage among LS carriers and can induce IFN-γ inflammatory responses in LS-derived TILs, offering an opportunity for vaccine development.
- Lynch Syndrome
- Dendritic
- Vaccine
- Immunotherapy
Data availability statement
Data are available in a public, open access repository. Data available on: Bayó Llorens, Cristina (2024). Supplementary information. figshare. Dataset. https://doi.org/10.6084/m9.figshare.27969414.v3 Data and study materials will be made available upon request to the corresponding authors. Public data from the study HNPCC-Sys: Molecular Characterization of Lynch Syndromes can be accessed under request on dbGaP repository accession number phs001407.v1.p1. Supplementary tables available under Figshare:53 Bayó Llorens, Cristina (2024).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Lynch syndrome (LS) is the most common hereditary form of colorectal and endometrial cancer, caused by germline pathogenic variants in DNA mismatch repair (MMR) genes.
Microsatellite instability in LS tumors generates highly immunogenic frameshift-derived neopeptides (FSDNs), making them targets for immunotherapy approaches.
Cancer vaccines targeting FSDN offer a promising approach to addressing the gap in cancer prevention for LS-associated tumors.
However, detailed information on targetable FSDN in the full spectrum of LS tumors and precursor lesions remains limited.
WHAT THIS STUDY ADDS
Identification and experimental validation of LS-related Human Leukocyte Antigen (HLA) class I and II-restricted FSDNs for cancer vaccine development in LS carriers.
The proposed approach circumvents the need for extensive mutation screening by targeting diverse neoantigens with varying HLA binding affinities to prevent immune evasion.
Immune responses against a wide range of predicted FSDNs have been detected on LS T cells from both precancers and cancers.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Provides a foundation for the development of off-the-shelf FSDN vaccines to enhance immune surveillance and prevent LS-associated tumors.
Offers insights into immunogenic FSDNs that may be adapted for sporadic MMR-deficient cancers, broadening implications for cancer prevention and immunotherapy.
Supports the design of clinical trials aimed at assessing the efficacy of FSDN-based vaccines in LS carriers.
Introduction
Lynch syndrome (LS) is the most common form of hereditary colorectal (CRC) and endometrial cancer (EC) accounting for up to 3–5% of all cases,1 2 with an estimated population-based prevalence of 1 in 279 individuals.3 It is an autosomal dominant syndrome due to a germline pathogenic variant in one of the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6 and PMS2) or deletions in the EPCAM gene.2 4 LS predisposes to the development of different tumors, mainly CRC (accumulated risk of 12–48%) and EC (13–49%),5 6 although differences in penetrance and age of onset vary on the affected gene. The tumor spectrum is much broader (stomach, urinary tract, ovary, small intestine, bile ducts, skin, brain), however, these neoplasms are much less frequent.
LS tumors are characterized by dysfunction of the MMR system (mismatch repair deficient (MMRd)) due to inactivation of both alleles of the MMR genes. MMRd cells accumulate numerous somatic mutations, including insertion/deletion (indel) mutations predominantly in repetitive sequences called microsatellites (MS)7 thus leading to a phenotype called microsatellite instability (MSI). Indels occurring in coding MS promote translational frameshifts which can generate frameshift-derived neopeptides (FSDN) that have the following features: (1) they are usually highly immunogenic (MMRd tumors are among the most immune-infiltrated and responsive to immune checkpoint inhibitors8–10; (2) there are many shared FSDN among patients and tumors from LS carriers11 12; and (3) they constitute “ideal targets” for immune interception because they have not generated prior immune tolerance. The existence of shared FSDN in MMR-deficient cancers creates a mechanism-based framework for novel tumor immunopreventive approaches,13 14 where several studies have already identified a large spectrum of genes affected by such frameshift mutations.11 Due to this “mutator phenotype”, the current hypothesis is that LS individuals are under constant immune surveillance and T cells specific to several FSDNs can be found in blood, healthy mucosa, precursor lesions such as colorectal adenomas, and early-stage cancers.15–17
Intensive surveillance colonoscopy is effective in decreasing CRC incidence; however, some CRCs escape prevention, and no preventive strategies exist for the majority of LS-related tumors. Vaccination with FSDNs that are shared by multiple MMR-deficient tumors is a promising approach to boost immune surveillance of MMR-deficient precancerous cell clones.13 Vaccination against only four FSDNs has shown to prevent tumors in a mouse model using VillinCreMsh2 mice, which have a conditional knockout of Msh2 in the intestinal tract and develop intestinal cancer.18 In humans, a phase IIa clinical trial in patients with a personal history of MMR-deficient CRC demonstrated the safety and immunologic efficacy of a trivalent recurrent FSDN-based peptide vaccine.19 However, identification of the most effective FSDN and whether vaccination can reduce LS tumor burden, in addition to boosting antitumor immunity, is still being studied.15
Many computational algorithms and machine-learning tools are available for identifying and prioritizing mutations that lead to FSDN that are likely to be recognized by T cells.20 Most trials focus on major histocompatibility complex (MHC)-I restricted predictions due to the limited accuracy of current MHC-II prediction algorithms. However, studies have shown that the expression of a single MHC-I neoantigen alone is insufficient; at least one additional MHC-II neoantigen is required for meaningful antitumor immunity.21 This underscores the importance of CD4+T cell activation in achieving a comprehensive antitumoral response.
In this study, we conducted a bioinformatic prediction and validation of LS-related Human Leukocyte Antigen (HLA) class I and II-restricted FSDNs. We found a set of neoantigens with high prevalence on LS and MMRd tumors and precancers, which induced interferon (IFN)-γ-producing pro-inflammatory responses in normal mucosa, tumor and adenoma-infiltrating T-lymphocytes from patients with LS. These FSDNs will be included in a semi—“off-the-shelf” autologous dendritic cell-based vaccine aimed at preventing cancer in LS.
Methods
A summary of the methodology used for this study is represented in figure 1. This study was conducted in three phases. In the first phase, an in silico prediction and prioritization of FSDNs in LS neoplastic lesions was carried out based on the most common mutations in coding MS. In the second phase, the presence of mutations associated with these FSDNs was validated using public databases and sequencing of colorectal adenoma (CrAD) and EC from individuals with LS. Finally, in the last phase, the antigenic recognition of the prioritized FSDNs (pFSDNs) was validated in colon samples from LS carriers.

Summary of the methods for bioinformatic FSDN prediction and validation. A first phase of bioinformatic FSDN prediction was carried out by using the pVACbind pipeline on commonly mutated coding MS sequences in MSI tumors. Once an optimal pFSDN set was filtered out, an in silico validation was performed by searching for the prioritized FSDN-associated mutations in DNA sequences from LS-related tumors, sporadic MMRd, as well as DNA and RNA sequences from human tumorous cell lines. A final phase of in vitro analysis was carried out to determine which of the pFSDN induced an inflammatory response on patients with LS’s mucosa infiltrating T lymphocytes. CCR, Colorectal Cancer; CrAD, colorectal adenomas; CRC, colorectal cancer; EC, endometrial cancer; ELISpot, enzyme-linked immunospot; FSDN, frameshift-derived neopeptides; HCB, Hospital Clinic of Barcelona; HLA, Human Leukocyte Antigen; iDCs: induced dendritic cells; IFN, interferon; LS, Lynch syndrome; mDC, mature dendritic cells; MMRd, mismatch repair deficient; MSI, microsatellite instability; MS, microsatellites; NMD, non-mediated decay; pFSDN, prioritized FSDN; TCGA, The Cancer Genome Atlas; TIL, tissue-infiltrating lymphocytes.
Participants and samples
Formalin-fixed paraffin-embedded (FFPE) samples from 17 advanced CrADs and 8 EC from 8 patients with LS were obtained from the Gastroenterology and Gynecology Department of Hospital Clinic of Barcelona (HCB) (online supplemental table 1). All CrADs and ECs displayed MMR deficiency by immunohistochemistry, showing loss of MMR protein expression. 23 additional individuals (8 for the first in vitro studies and 15 for the following ones) with LS were recruited from the Gastroenterology Department of the HCB (online supplemental tables 2 and 12, respectively) for the in vitro validation of the pFSDN. During surveillance colonoscopy, 40 mL of peripheral blood and biopsies of normal mucosa, CrADs or CRCs from each individual were collected. HLA haplotype of all the participants was determined using high-resolution next-generation sequencing (NGS) typing for the HLA-A, HLA-B, HLA-C, HLA-DR1, HLA-DQ1, and HLA-DP1 loci in the molecular biology Core from the HCB.
Supplemental material
In silico FSDN prediction and prioritization
Selective Targets in Human MSI-High Tumorigenesis Database (Seltarbase, www.seltarbase.org22,22 accessed September of 2021) and different bibliography sources14 23 were used to identify FSDN in coding MS from MSI-high tumors. As the Seltarbase database is no longer available and cannot be accessed, we provide the initial sequences selected from all sources used in online supplemental table 3. We included coding MS with a length of at least eight bases, coding MS in putative tumor-driver genes and oncogenes (according to recent standardized lists of potential cancer driver genes24–26) and coding MS known to produce frameshift peptides with predicted high-affinity ligands according to literature. 531 frameshift peptide sequences derived from m1 (deletion of 1 nucleotide) and m2 (deletions of 2 nucleotides) mutations in 269 coding MS were identified and processed through the pVACbind prediction pipeline (pVACtools V.3.1.027) for FSDN prediction against the 40 most common HLA class I (n=30) and II (n=10) alleles in the European population (online supplemental figure 1).28 This pipeline does not require Whole Exome Sequencing (WES) and RNA sequencing data to predict FSDN, which we did not use due to the unavailability of sufficient samples for a generalized output.
Supplemental material
The output results of the pVACbind pipeline were classified as strong (IC50: <50 nM), weak (IC50: 50–500 nM) and very weak (IC50: 500–5000 nM) affinity binders to HLA and filtered by the criteria stated in the supplementary methods (online supplemental methods S1-3, online supplemental figure 3, online supplemental table 4, online supplemental table 5).
Supplemental material
Validation of prioritized FSDN using public databases and direct sequencing of LS neoplasia
DNA sequences from The Cancer Genome Atlas (TCGA) (CrAD, CRC from LS and sporadic MMRd cancers).
We obtained access to whole genome sequencing (WGS) data from the National Institutes of Health study HNPCC-Sys: molecular characterization of LS (phs001407.v1.p1; dbGaP, TCGA) carried out by the HNPCC-Sys consortium. This study was supported by the German Federal Ministry of Education and Research with the PTJ grant HNPCC-Sys 031 6065A. The study gave access to 98 samples from CrADs (n=10), CRC (n=40), and paired healthy samples (n=48) of 11 patients with LS (online supplemental table 6). The FASTQ files obtained for WGS sequences from HNPCC-Sys were transferred to the Barcelona Supercomputing Center central storage for read pre-processing and quality control (online supplemental methods S4). We used three different somatic variant callers Mutect,29 SAMtools30 and VarScan,31 and ran each of them using the default parameters. The Variant Effect Predictor (VEP) tool32 has been used for the annotation and prioritization of genomic variants in coding and non-coding regions. Only the variants annotated as frameshift and common to all three variant callers were considered in the following analysis.
Additionally, we analyzed the pFSDN-associated mutations in sporadic MMRd cancers. Annotated frameshift variant files from MSI-high (MSI-H), MSI-low (MSI-l) and microsatellite-stable (MSS) exome sequencing samples were obtained from different projects with high prevalence of MSI in the TCGA database (genomics data portal database, National Cancer Institute; NCI), including TCGA-BRCA (breast cancer, n=15), TCGA-CESC (cervix uteri cancer, n=8), TCGA-COAD (colorectal adenocarcinoma, n=84), TCGA-READ (rectum neoplasms and adenomas, n=11), TCGA-STAD (stomach cancer, n=84) and TCGA-UCEC (corpus uteri cancer, n=164) (online supplemental table 7), either by direct download from their domain or from the study in reference.33 Status of MSI, if not provided by the source, was established by the MSI detection tool MANTIS.34 The annotated variant files from human sporadic MMRd tumor sequences were converted from GChr38 to GChr37 using the LiftOver conversion tool from the UCSC Genome Browser. We then identified the presence of pFSDN-associated mutations in these sequences by comparing the genome coordinates of each pFSDN-associated mutations with the retrieved sequence files in GChr37 format.
Targeted MS sequencing of CrAD and EC samples from patients with LS cancer
Highly sensitive microsatellite instability (hs-MSI) assessment of 17 CrAD and 8 EC samples from patients with LS was performed by using a custom panel targeting 192 microsatellites using HaloPlex HS technology (Agilent Technologies) as previously described.35 36 The hs-MSI panel included 93 coding MS, 27 of which matched our pFSDNs (corresponding to 40 pFSDN). Library preparation and bioinformatics analysis are detailed in online supplemental methods S5. The obtained targeted sequence data were computed through a high-sensitivity software for the detection of insertions and deletions.35 The variant calling files from patients with CrAD and EC were annotated with the Ensemble VEP tool and were subsequently used for the pVACseq neoantigen prediction analysis to predict the binding of FSDN to the HLA alleles of each patient. The program was run with default settings and with the predictors MHCnuggetsI, MHCnuggetsII, NetMHC, NetMHCcons, NetMHCIIpan, NetMHCpan, NNalign and PickPocket. The resulting files were filtered to exclude neoantigens with an HLA binding threshold greater than an IC50 of 5,000 nM.
Genomic and transcriptomic data from MSI cell lines
We selected MSI cell lines with available variant WES calling files at the Cell Model Passports website (https://cellmodelpassports.sanger.ac.uk; V.2.7.0) from the Sanger Institute37 (online supplemental table 8). RNA sequencing (Illumina 2000 or 6000) data was retrieved from 11 of them to analyze messenger RNA (mRNA) expression. DNA data was accessed through the public database Cell Model Passports, whereas RNA data has been downloaded from the Sequence Read Archive (SRA) from different groups that posted cell line transcriptomics information. DNA and RNA sequencing data analyses are detailed in online supplemental methods S6.
In vitro validation of prioritized FSDN in LS colorectal neoplasia from LS carriers
The top 98 pFSDN obtained from in silico validations were synthesized by Proteogenix (France) at >80% purity and 4 mg per peptide. Synthetic pFSDNs were reconstituted with either dimethyl sulfoxide (DMSO) (Werfen, Spain) or acetonitrile (Sigma-Aldrich, Germany): H2O (1:3), following providers’ recommendations, at a stock concentration of 5 mM.
FSDN cytotoxicity assessment on dendritic cells
Monocyte-derived dendritic cells (DCs) (mo-DCs) were generated from peripheral blood mononuclear cells (PBMCs) from five healthy controls and eight patients with LS as stated in online supplemental methods S7. mo-DCs from healthy donors were used to assess FSDN cytotoxicity on these cells by loading them with FSDN pools and studying (1) differences on maturation markers and (2) their capacity to stimulate proliferation on allogenic T cells by a mixed lymphocyte reaction (MLR) (online supplemental methods S8–S9).
Tissue-infiltrating lymphocytes and mo-DCs co-cultures for bulk IFN-γ enzyme-linked immunospot
Matched LS CrADs or CRC biopsies were processed for tissue-infiltrating lymphocytes (TILs) isolation and expansion as stated in online supplemental methods S10–S11. We decided to use TILs instead of PBMCs as we expect a higher response on mucosa than in peripheral blood. Cryopreserved LS mo-DCs were thawed, matured in a pro-inflammatory environment, and loaded with the 98 synthetic peptides individually. Autologous T cells from patients with LS were co-cultured with LS-loaded mature mo-DCs in a 1:10 ratio on IFN-γ enzyme-linked immunospot (ELISpot) plates (Mabtech, Sweden). Positive controls used the kit’s CD3 stimulation, and negative controls used DMSO. Spot forming units (SFUs) were measured to assess the immune response. Tests were considered positive when the number of spots was 1.5 times higher than the negative control. We used only a 1.5-fold threshold for ELISpot positivity due to an expected high background noise from the co-culture of non-loaded mature mo-DCs plus autologous TILs, which usually induces an unspecific pro-inflammatory stimulus. See online supplemental methods S12.
Neoantigen-specific T cell expansion using a set of prioritized FSDN
Based on data from both in silico and in vitro bulk analyses, we prioritized a refined peptide set of 18 HLA-I and 6 HLA-II restricted peptides. Prioritization was based on peptides meeting the following criteria: (1) a positive ELISpot antigenic response, (2) lack of sequence similarity to self-proteins, (3) broad HLA allele coverage, and (4) high predicted occurrence in LS-related or sporadic MMRd tumors (online supplemental table 10). Additionally, three peptides (MSH3, TGFBRII_B, and ACVR2A_II) were included based on their strong immunogenicity in clinical trials (NCT01885702) and were generously provided by Dr Beatriz Carreño (University of Pennsylvania) and Dr Jolanda M De Vries (Radboud Institute for Molecular Life Sciences).
CrAD or normal mucosa biopsies and 40 mL of blood were collected from 13 additional LS carriers. PBMCs were isolated from whole blood by Ficoll gradient separation (STEMCELL technologies, Grenoble, France) and biopsy-derived TILs were obtained as a cell suspension (CS) (online supplemental methods S10). PBMCs were irradiated (40 Gy, 30 min) and co-cultured with CS at a 1:5 ratio (CS:PBMCs) in T-cell media without interleukin (IL)-2, with at least 100,000 CS cells per well across five wells of a p48 plate. The HLA-I peptide pool (5 µM/peptide) was added, and cells were incubated for 48–96 hours before IL-2 (20–40 U/mL) supplementation. Media were refreshed every 48 hours until day 14, when T cells were re-stimulated with frozen PBMCs (1:5 ratio). Cells were tested for HLA-I peptide pool reactivity via IFN-γ ELISpot (Thermo Fisher) at day 14 and 28. Each well contained 50,000–100,000 cells in duplicates, with two negative controls (DMSO and an irrelevant peptide at 5 µM). As the expected background for these analyses was low and we were interested in detecting robust IFN-γ responses, the positivity threshold was established at ≥3× the negative control. Post-ELISpot, cells were pooled and analyzed by flow cytometry using monoclonal antibodies (CD3-FITC (Clone HIT3a), CD4-V500 (Clone RPA-T4), CD8-APC (Clone RPA-T8), CD154-BV421 (Clone TRAP1), CD137-PE (Clone 4B4-1), CD69-APC-Cy7 (Clone FN50) (BD, Spain)), on an Attune NXT cytometer. Phycoerythrin (PE)-labeled pentamers for selected HLA-I-peptide combinations (Proimmune, UK) were used per manufacturer instructions.
Statistical analyses
Statistical tests were performed with GraphPad Prism V.8.0.1. The Shapiro-Wilk test was performed to determine normality, and paired t-tests (for Gaussian distributed cohorts) or Kolmogorov-Smirnov test (for non-Gaussian cohorts), Brown-Forsythe and Welch’s analysis of variance (ANOVA) or ordinary one-way ANOVA were used to determine significance when comparing cohorts. Quantitative results are shown as median (IQR). Significance shown in American Psychological Association style (p value: “.123” ns, “.012” *, “.001” **, “<0.001” ***).
Results
In silico prediction, characterization and prioritization of shared FSDN in MSI tumors
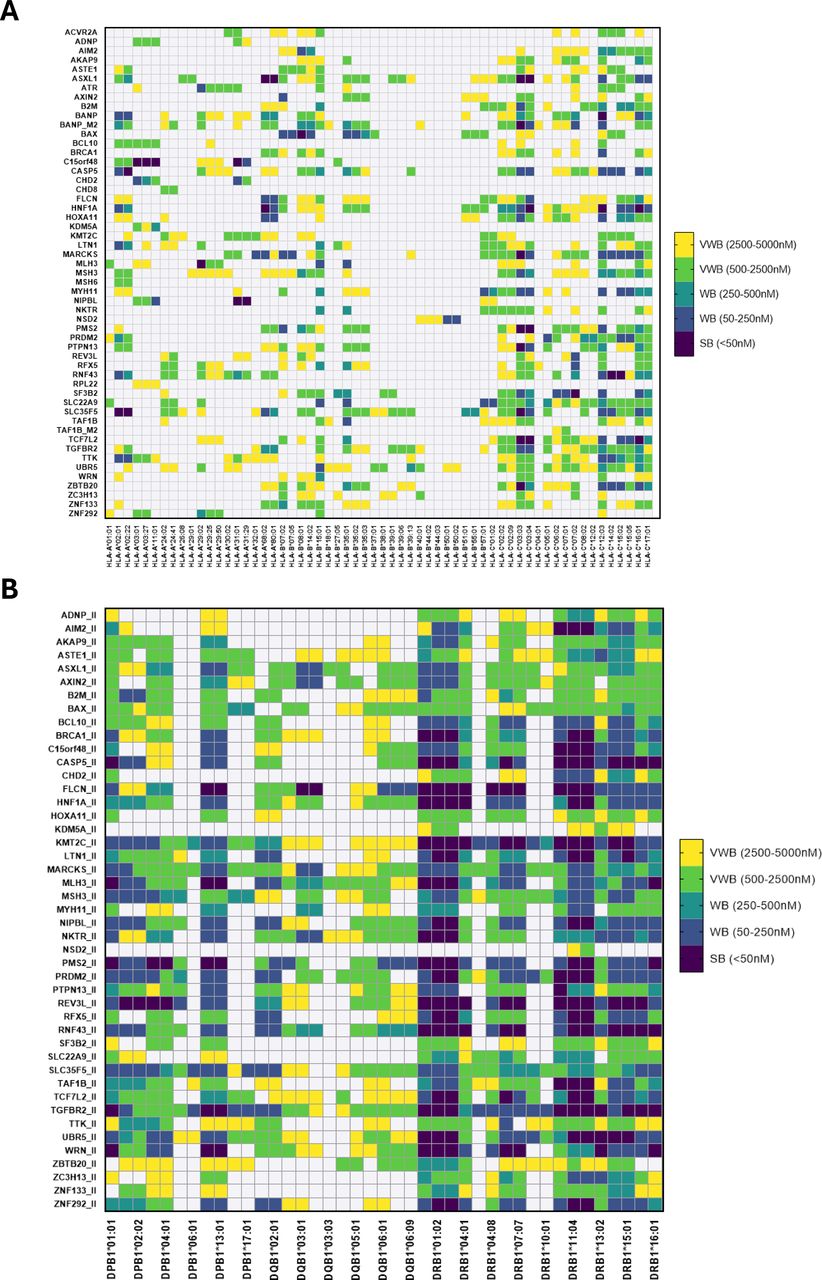
To predict shared neoantigens among different LS and MMR deficient tumors we used coding MS sequences derived from m1 (deletion of 1 nucleotide) and m2 (deletions of 2 nucleotides). In total, 42,828 HLA-I-restricted and 8,350 HLA-II-restricted epitopes were identified, which were filtered and reduced to 53 FSDN restricted in length to HLA-I and 45 to HLA-II (figure 1), which derived from a total of 53 different frameshift mutations. These pFSDNs were classified as strong, weak and very weak affinity binders to HLA molecules (figure 2). The final set of genes associated with the pFSDN is detailed in online supplemental table 9.

Prioritized frameshift-derived neopeptides binding affinity to HLA-I (A) and HLA-II (B) tested alleles. Epitopes are classified and color-coded in strong (IC50<50 nM), weak (tier 50 nM<IC50<250 nM and tier 250 nM<IC50<500 nM), and very weak (tier 500 nM<IC50<2500 nM and tier 2500 nM<IC50<5000 nM) binders. HLA, Human Leukocyte Antigen; SB, strong binders; WB, weak binders; VWB, very weak binders.
For further characterization of the pFSDN, we carried out a self-similarity analysis against the human proteome to assess the possibility of generating autoimmunity. Results determined that eight of our pFSDN (8.2%) had significant similarity to a self-protein (online supplemental table 4). We also performed a prediction of sequence similarity to protein sequences from known human pathogens, which has been demonstrated to increase the immune response generated against neoepitopes due to cross-reactivity by molecular mimicry. 22 pFSDN (22.4%) were reported to have protein sequence similarity to at least one of the human infectious pathogens’ peptide sequences with positive immune assays analyzed (online supplemental figure 2, online supplemental table 5), of which 40% had similarities to more than one pathogenic sequence. Lastly, 44.2% of all pFSDN were predicted to be resistant to non-sense-mediated RNA decay, a mechanism that degrades non-sense mRNA usually derived from frameshift mutations that give rise to immunogenic neopeptides. Resistance to this process increases the probability of epitopes being presented in MMRd cells.
Validation of the prioritized FSDN-associated mutations in LS CrAD, CRC and EC samples and sporadic MMRd tumors
To validate the presence of the set of pFSDN, we searched for their associated mutations in different cancers and CrADs from different sources.
Lynch syndrome CrADs and CRC (HNPCC-Sys data)
The mutational pattern of WGS data from the study HNPCC-Sys revealed a high proportion of frameshift deletions in mononucleotide repeat sequences in both CrAD and CRC samples (figure 3 and online supplemental figure 4 A,B and C). The average number of variants and indel (median(IQR)) variants per sample was increased in CrADs (101.5, 60.5 (21.13–69.04)) and in CRCs (50.5, 17.1 (11.13–40.17) samples compared with paired control samples (38.5, 6.5 (3.58–9.10)). No significant differences were found in the variant allele frequency (VAF) of these mutations across sample types. Focusing on our list of HLA-class I pFSDN associated mutations, we found that 46 (86.7%) out of 53 were present on CRC samples. Specifically, TGFBR2, SLC35F5, ACVR2A, ASTE1, TTK and ZNF292 were mutated in >25% of CRC samples, and TGFBR2, SLC35F5, ACVR2A, ASTE1, LTN1 and RPL22 were mutated in >35% of CrAD samples. The median of pFSDN-associated mutations per sample type was 3.05 (1–4) in control paired normal mucosa samples, 7.67 (6.5–9) in CrAD samples and 6.02 (2–10) in CRC samples. Interestingly, MLH1/MSH2 pathogenic variant carriers’ samples were associated with a higher number of pFSDN associated mutations compared with MSH6 carriers (5.39 (2–8) vs 2.11 (1–3.5), respectively; p=0.0351*). Due to HLA typing data being unavailable for the patients included in this study, FSDN binding predictions could not be performed.

Occurrence of prioritized FSDN associated mutations in lynch syndrome samples from the study HNPCC-Sys. (A) Oncoplot depicting the occurrence of prioritized FSDN associated mutations in control, CrAD and colorectal cancer samples. Top panel shows the number of variants detected in each sample. The right panel shows a heatmap (bottom) and a boxplot (top) of the relative percentages of mutated genes in control, CrAD and tumor samples. (B) Boxplot (mean with SD and 95% percentile) showing the number of pFSDN associated mutations per sample by germline mismatch repair deficient mutations. Unpaired t-test was performed to determine significance (p value in American Psychological Association style: “.123” ns, “0.012” *, “.001” **, “<0.001” ***). CrAD, colorectal adenomas; FSDN, frameshift-derived neopeptides; pFSDN, prioritized FSDN.
Sporadic MMRd tumors (TCGA data)
We next explored whether pFSDN-associated mutations were highly ranked in recurrently targeted MS loci and whether they were specific to MSI-H tumors. We examined the top 1,000 most recurrently altered MS loci from 190 MSI-H and 640 MSS exomes in TCGA data sets (COAD, UCEC, and STAD)33 by comparing mutational coordinates in coding Mononucleotide Repeat (MNR) sequences. As expected, we found that MSS and MSI-L samples had little to no pFSDN-associated mutations (mean 0.2%±0.24 samples per mutation), while MSI-H samples showed a significantly higher prevalence (mean 23.15%±14.8 samples per mutation) (figure 4A and B). We observed that 22 of our pFSDN mutations (41.5%) were highly ranked among the most altered MS loci in non-LS MMRd MSI-H tumors. A deeper analysis from mutations in MSI-H sequences independent of their recurrence in MS loci from additional TCGA data sets (COAD, UCEC, STAD, READ, CESC, and BRCA) demonstrated that 48 out of 53 (90.76%) pFSDN-associated mutations were present in the analyzed sequences, each appearing in a median of 16±33.03 samples (figure 4C). In TCGA-CESC, COAD, STAD, and UCEC, over 55% of samples contained pFSDN-associated mutations, with UCEC showing the highest representation (80.35% of samples, n=168) and READ the lowest (18.18% of samples, n=11). Among all pFSDN mutations, ACVR2A, RPL22, SLC22A9, ASTE1, RNF43, TGFBR2, and ZBTB20 were the most frequently mutated, appearing in over 25% of samples.

Prioritized FSDN-associated mutations’ presence in non-LS MSS and MSI-H samples. Prioritized FSDN-associated mutations in coding microsatellites from TCGA data sets colon adenocarcinoma (COAD), uterine corpus endometrial carcinoma (UCEC), rectum adenocarcinoma (READ), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), stomach adenocarcinoma (STAD), and breast invasive carcinoma (BRCA). (A) Heatmap representing the relative percentage of samples containing the pFSDNm in the top 1,000 altered coding microsatellites from TCGA data sets categorized by MSI-H or MSS status. COAD, UCEC and STAD. (B) Scatterplot (mean with SD) showing the differences in prioritized FSDN-associated mutations occurrence between MSI-H and MSS samples from COAD, UCEC and STAD data sets. Significance determined by t-test (p value in American Psychological Association style: “.123” ns, “0.012” *, “.001” **, “<0.001” ***). (C) Oncoplot showing the occurrences of prioritized FSDN associated mutations on TCGA data sets. The frequency (percentages on the left and right barplot) is calculated by dividing the number of variants in the specific gene by the number of patients with at least one mutation identified. Top panel shows the number of aberrations detected in each sample. The heatmap on the right panel shows the relative percentages of prioritized FSDN-associated mutations. FSDN, frameshift-derived neopeptides; MSI-H, microsatellite instability-high; MSS, microsatellite-stable; pFSDN, prioritized FSDN; pFSDNm, prioritized FSDN mutations; TCGA, The Cancer Genome Atlas.
Lynch syndrome CrADs and EC (NGS sequencing)
Sequencing FFPE samples from 17 CrADs and 8 EC (online supplemental table 2) from patients with LS was performed using an NGS-high sensitivity MSI panel covering 27 of the selected coding MS corresponding to 40 pFSDN. All samples had MSI scores consistent with MSI-H lesions. The resulting reads had a mean coverage >932 and a minimum average of unique reads of 84.1%. Studying their mutational patterns revealed that 71% of the coding MS readings had m1 deletions and 1% had 2p insertions, which alter the reading frame to M1 (72%) (figure 5A). Most coding MS alterations favor a frameshift to the M1 reading frame, which corresponds to the origin of 97% of our pFSDN associated variants. Mutational prevalence analysis showed that all CrAD and EC samples harbored pFSDN-associated mutations, where 72.5% of them were shared among both sample types (online supplemental figure 5A). Notably, MARCKS, SLC35F5, TGFBR2, and BAX variants appeared in over 50% of both CrAD and EC samples, underscoring the high prevalence of these shared mutations. Additionally, ASTE1, KDM5A, AIM2, MSH3, URB5, and CASP5 mutations were frequently observed in more than 50% of CrAD samples (figure 5B). Interestingly, MLH1 and MSH2 pathogenic variant carriers had a significantly higher occurrence of pFSDN-associated mutations in CrAD and EC compared with those with MSH6 variants (p=0.0234) (figure 5C).

Analysis of LS-related colorectal adenoma (n=17) and endometrial cancer (n=8) samples. (A) Pie chart showing the distribution of all coding MS frameshift mutations (minus (m) 5 – plus (p) 2) and their corresponding frames (M0, black; M1, aquamarine; M2, beige). (B) Mutational profile of LS EC (bottom) and CrAD (top) samples from coding MS targeted DNA sequencing classified by the mutation type (1 bp deletion or 2 bp insertion, M1; 2 bp deletion or 1 bp insertion, M2; synonymous mutation, (M3). C) Barplots (mean with SD) showing the number of frameshift deletions (left), insertions (center) and number of predicted prioritized FSDN expression (right) are represented by germline mismatch repair deficient mutations. Unpaired t-tests were performed to determine significance (p value in American Psychological Association style: “.123” ns, “0.012” *, “.001” **, “<0.001” ***). (D) Analysis of LS-related colorectal adenoma (n=17) and endometrial cancer (n=8) samples. Heatmap of the pVACseq neoepitope prediction against each patient HLA haplotype. The color code corresponds to the IC50 values for the strongest HLA allele binder from each patient classified as strong (SB), weak (WB), and very weak (VWB) binders. CrAD, colorectal Adenomas; EC, endometrial cancer; FSDN, frameshift-derived neopeptides; HLA, Human Leukocyte Antigen); LS, Lynch syndrome; MS, microsatellites.
Computing sequenced data through the pVACseq neoantigen prediction software against each patient’s HLA haplotype determined that 38/40 (95%) pFSDN had a high predicted probability of being presented on HLA-I or II molecules in CrAD samples, and 31/40 (77.5%) in ECs, with a median of 4.92 (2–7) predicted strong binding (<100 nM) (3.625 (2–4.5) for ECs and 5.53 (3–8) for CrADs) and 9.96 (5–14) mid to very weak binding (100–5000 nM) (5.5 (2.5–8.5) for ECs and 12.06 (6–17) for CrADs) pFSDN per sample. 24 out of 40 (60%) pFSDNs appeared in ≥50% of CrAD samples and 12 (30%) in ≥50% of EC samples. pFSDNs restricted to class I and II derived from MARCKS, SLC35F5, TGFBR2 and BAX were the most prevalent considering both sample types (figure 5D).
MSI cancer cell lines
We next analyzed the presence of our pFSDN-associated variants and corresponding mRNA transcripts in MSI CRC and EC confidence levels (CLs). This analysis provides crucial information on whether the analyzed and pFSDN-associated mutations are specific to an MSI phenotype and transcribed into mRNA in an in vitro model (online supplemental table 8). Cell lines from ECs and CRCs with MSI harbored up to 35 out of the 53 (66%) pFSDN associated variants in DNA sequences, with a median of 8.88 (7–10.25) mutations per MSI CRC CL and 7.1 (3–10.5) per MSI EC CL. No statistically significant differences were observed among the number of pFSDN associated mutations between CRC and EC MSI CLs, but the correlation and trend line between sample types was significant (p value=0.0002441) (online supplemental figure 6). Despite this, we found that 65.7% of the pFSDN associated mutations were shared between both sample types (online supplemental figure 5A). When analyzing the mRNA sequences, 37 out of the 53 (69.8%) pFSDN associated mutations were transcribed in MSI cell lines with a high VAF (median of 0.53 (0.264–0.741)) (online supplemental figure 6). Altogether, this analysis demonstrates that our pFSDN associated mutations are transcribed to RNA in MSI cell lines.
FSDNs elicit IFN-γ responses in LS tissue-infiltrating lymphocytes
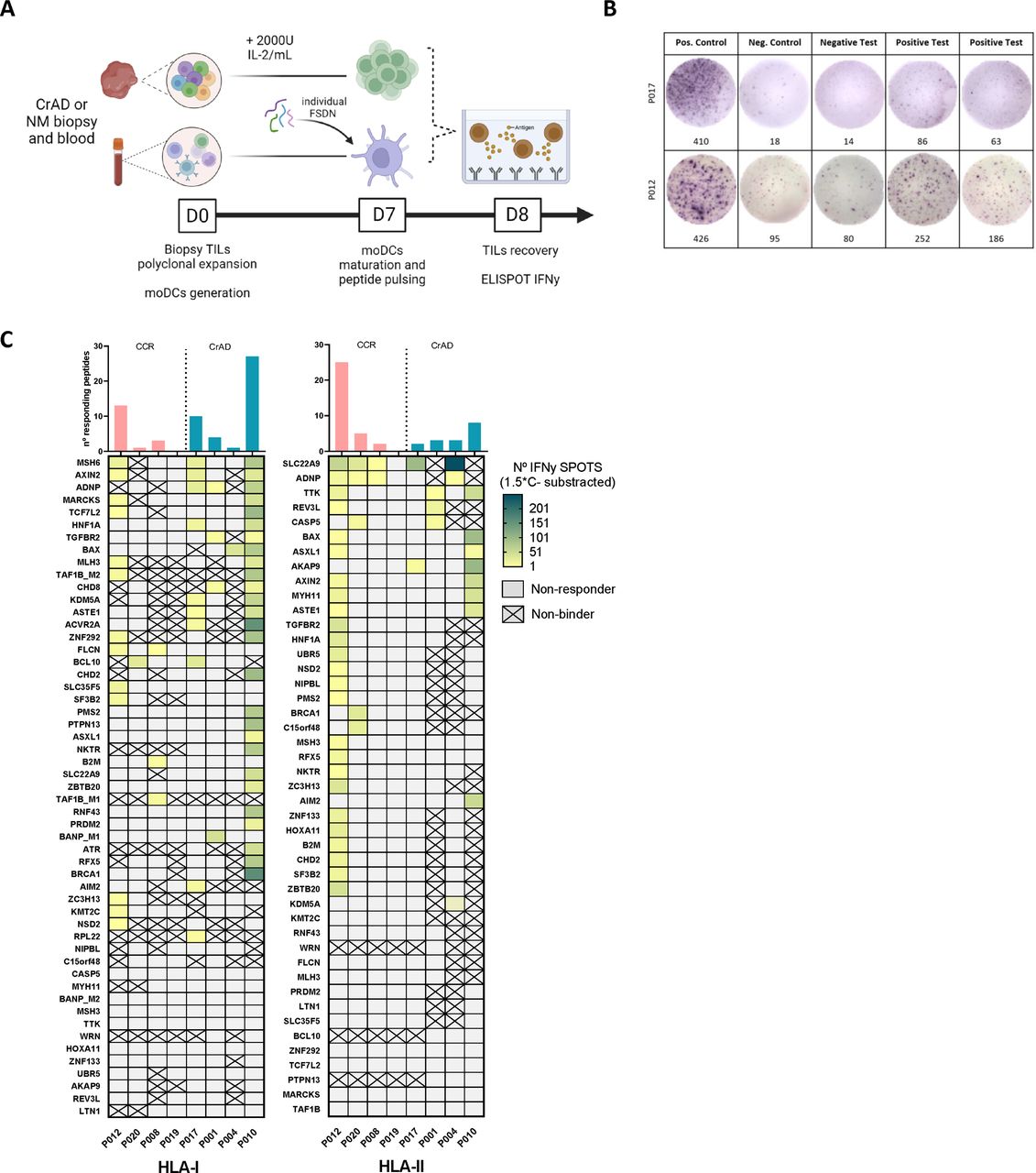
One of the primary challenges of FSDN discovery is accurately identifying peptides capable of eliciting a robust immunogenic response. As a first approach to entirely discard those peptides from our initial FSDN set that were not recognized or that do not induce an IFN-γ response in any patients with LS, we stimulated TILs from CRCs and CrADs from patients with LS using synthetic pFSDNs loaded on autologous mo-DCs. We then quantified IFN-γ release using ELISpot assays. The characteristics from the included patients in this first assessment are summarized in online supplemental table 2.
The heatmap in figure 6C shows IFN-γ ELISpot results (SFUs/peptide) which varied greatly among patients. These differences likely reflect the hypermutator phenotype of patients with LS and different HLA haplotypes. Our analysis revealed that 70 out of 98 (71%) pFSDNs elicited a positive IFN-γ ELISpot response (exemplified in figure 6B), where 7 out of 8 (87.5%) patients responded to FSDNs, with a median of 7.37 (1–10.75) responding HLA-I pFSDN per patient (10.5 (3.25–14.25) in CrADs and 4.25 (0.75–5.5) in CRCs), and 6 (2–5.75) for HLA-II pFSDNs (4 (2.75–4.25) in CrADs and 8 (1.5–10) in CRCs).

IFN-γ ELISpots in response to synthetic pFSDNs. (A) Schematic representation of the protocol used. (B) Random examples of IFN-γ ELISpots positive control, negative control, positive test and negative test wells. (C) Heatmap depicts the number of spots per peptide (y axis) and patient (x axis) after subtraction of the negative control, separated by HLA-I restricted pFSDNs (left panel) and HLA-II restricted (right panel). Cells defined as “non-responder” in gray represent negative ELISpots for peptides predicted to bind to any HLA alleles from the corresponding patient (IC50<5000 nM), whereas crossed-out cells defined as “non-Binder” correspond to negative ELISpots for pFSDNs without any predicted compatible HLA alleles in the corresponding patient (IC50>5000 nM). Barplots on top of the plots represent the total number of peptides that induce a positive IFN-γ response for each patient. CrAD, colorectal adenomas; ELISpot, enzyme-linked immunospot; FSDN, frameshift-derived neopeptides; HLA, Human Leukocyte Antigen; IFN, interferon; IL, interleukin; NM, normal mucosa; pFSDN, prioritized FSDN; TIL, tissue-infiltrating lymphocyte.
FSDNs do not affect DCs viability and maturation capacity
To determine possible FSDN cytotoxicity to mo-DCs, we generated and loaded mature moDCs from five healthy donors, phenotyped them and assessed their capacity to stimulate T-cell proliferation by an MLR. All DCs had >85% viability, which did not decrease when loaded with FSDNs. HLA-I and HLA-DR (HLA-II molecule) were expressed by >85% in all conditions, CD80 expression was increased in all mature DCs (mDCs) (>70%) compared with induced DCs (iDCs) (mean 53% (39.45–62.18)), CD83 followed a similar pattern with >92% expression on all mDCs conditions and a mean of 48% (36.75–58.75) in iDCs, and CD86 remained with a high expression (>84%) in all conditions; however, a significant difference in the mean fluorescence intensity is observed on all mDCs conditions compared with iDCs (online supplemental figure 7A,B). With the MLR, we confirmed that all pulsed mDCs maintained the capacity of stimulating allogenic T cells, with a negative CFSE (Carboxyfluorescein succinimidyl ester)-stained population of >23% on average on all mature conditions compared with iDCs (mean 16.88% (7.8–30.3)) (online supplemental figure 7C).
Prioritized FSDN-specific T cells are found in normal mucosa and CrADs from LS individuals
We prioritized 24 peptides for in vitro studies including 18 HLA-I and 6 HLA-II restricted peptides. Population coverage analysis using the Immune Epitope Database analysis resource38 confirmed high predicted response rates (>90%) across Spanish, European, and global populations for both HLA-I and HLA-II epitopes (online supplemental table 11).
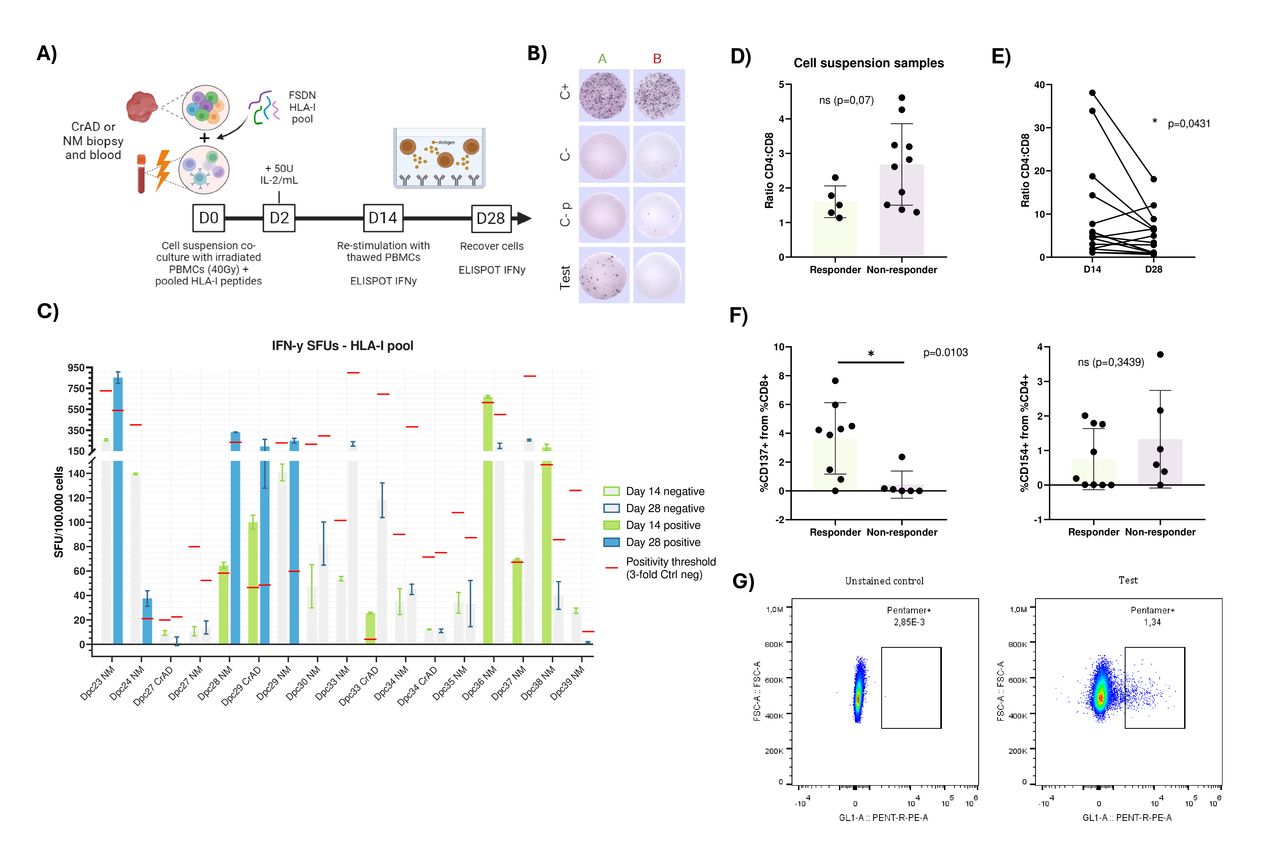
With the aim of detecting peptide-specific T cells, we tested the prioritized 18 HLA-I peptides by stimulating LS-derived healthy mucosa and CrAD-infiltrating T cells with autologous irradiated PBMCs, assessing responses via IFN-γ ELISpot and flow cytometry (figure 7A). A positive response was defined as a ≥3-fold increase over negative controls (exemplified in figure 7B). Notably, we applied a higher threshold for ELISpot positivity in these subsequent analyses to ensure robust detection of immunogenic responses. The characteristics of patients included in these experiments are summarized in online supplemental table 12.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HLA-I restricted prioritized neoepitopes immunogenic recognition by LS healthy mucosa and CrAD-derived T cells. (A) Schematic representation of the protocol used for the analysis. (B) IFN-γ ELISpot analysis criteria. Randomly picked representative IFN-γ ELISpot plate reading. Column A (green) shows a positive result (n° spots test ≥3 times n° spots C-), column B (red) shows a negative result. First row corresponds to the positive control, second row to the negative control (no added stimuli), third row to the negative control plus an irrelevant peptide at the same concentration as the ones added to test wells, and last row corresponds to the test wells. (C) IFN-γ ELISpots for the HLA-I peptide pool. withandBarplots showing SFUs per test, an n=2 technical duplicates with the SD are plotted, values are normalized for 100.000 cells. Two ELISPOT tests per patent are plotted for the first (Day 14, green) and second (Day 28, blue) stimulation with PBMCs and 5µM of the HLA-I peptide pool. Positive tests, with values above their threshold, are colour-filled, and negative tests bars remain grey. The positivity threshold for each ELISPOT, determined as 3-fold the negative control with an irrelevant peptide, is marked as a red line. (D) Comparison of CD4:CD8 ratio in responder and non-responder initial cell suspension samples, before cell expansion, from parent alive CD3+cells. Barplot shows n=14 independent experiments. (E) Evolution of CD8+cells percentage from expanded samples with the vaccine HLA-I pool, at day 14 and 28, from alive CD3+parent cells. Plot shows n=12 independent experiments. (F) CD8+CD137+ (left) and CD4+CD154+ (right) cells percentage in responder and non-responder samples after a 28-day expansion with the vaccine HLA-I pool. Plots show n=12 independent experiments. (G) Flow cytometry analysis of HLA-I DELAY-FSDN specific T cells using pentamers conjugated to PE for the peptides HLA-A*02:01-MSH3 and HLA-A*02:01-TGFBRII_B. Dot plots show cells gated from total alive CD8+cells. CrAD, colorectal adenomas; ELISpot, enzyme-linked immunospot; FSDN, frameshift-derived neopeptides; HLA, Human Leukocyte Antigen; IFN, interferon; IL, interleukin; NM, normal mucosa; PBMC, peripheral blood mononuclear cell; SFU, spot forming unit.
Among 17 samples (4 CrAD, 13 normal mucosa) from 13 patients with LS, 9 samples (53%) from 8 patients (62%) exhibited peptide-specific T-cell reactivity. Following a second PBMC stimulation, some samples (eg, Dpc23, Dpc24, Dpc28) showed increased specificity, while others (eg, Dpc36, Dpc38) lost reactivity due to non-specific overgrowth (figure 7C). Responder samples correlated with a higher CD8+proportion (p=0.07), and all samples showed increased CD8+percentages from day 14 to day 28 (p=0.0431). Only responder samples exhibited a significant CD8+CD137+ increase post-expansion (p=0.037), with no corresponding rise in CD4+CD154+ (figure 7D–F). In patient Dpc24, pentamer staining confirmed low-level HLA-A02:01-MSH3 and HLA-A02:01-TGFBRII_B-specific T cells, consistent with ELISpot results (figure 7G).
Discussion
In this study, we systematically identified LS-associated FSDNs using a custom filtering strategy. We validated the presence of FSDN-associated mutations across multiple data sets, including cancer and CrAD samples from LS carriers and sporadic MMRd patients. Our findings demonstrated that many of these mutations are transcribed into RNA in tumor cell lines and evade non-sense-mediated decay. Importantly, we showed that individuals with LS mount specific immune responses against these FSDN, as evidenced by IFN-γ ELISpot assays in infiltrating lymphocytes from normal mucosa, CrAD and CRC samples from both LS previvors and survivors. Our sequential approach has effectively identified a set of immunogenic peptides that could form the foundation of a cancer prevention vaccine.
This study highlights the importance of selecting neoantigens for LS prevention considering their cancer specificity, immunogenicity, and safety.19 39–42 We pFSDNs recognized by common HLA alleles in the European population to enhance the immune response across diverse individuals. By including both HLA class I and II epitopes, we ensured a comprehensive immune response involving both CD8+ and CD4+ T cells, crucial for effective tumor prevention and control.21 We further refined FSDN selection by filtering for self-similarity and pathogen-derived peptide similarity, enhancing cross-reactivity—22.4% of potential FSDNs showed similarity to pathogen peptides14 23 43—while minimizing the risk of autoimmunity by selecting peptides dissimilar to self, which is also associated with higher immunogenicity.44 Targeting neoantigens from driver mutations reduces immune evasion risks, focusing on clinically significant tumor cell populations, reducing the risk of immune evasion through antigen loss.45 Our analysis across data sets suggests these FSDNs are relevant not only for LS but also for sporadic MMRd tumors, indicating broader applicability of neoantigen-based vaccines in cancer prevention.15 46
Our approach is among the first to incorporate lower-affinity binding peptides—traditionally underexplored—while maintaining their immunogenic potential. Recent research shows that mid-affinity and low-affinity peptides play a crucial role in generating progenitor-like, multi-epitope targeting T cells that maintain immune surveillance. Incorporating peptides with varying HLA affinities could enhance immune vigilance.14 47–49 In this sense, even though our initial prediction of FSDN originated from cancer sequences, we included CrADs in our analysis, recognizing them as the main CRC precursor in LS. Recently, Bolivar et al published a study analyzing the mutational and neoantigen landscape of LS colorectal lesions, including CrADs.15 In that study, 65% of the top 100 predicted FSDNs were immunogenic in vitro and were present in the majority of CRCs as well as a significant proportion of CrADs. Their findings highlighted a significant number of MMRd CrADs bearing a high rate of neoantigens and immune activation. In our comparative analysis of sequenced ECs and crADs, despite inherent differences—such as CrADs being precursor lesions rather than fully developed cancers—we observed shared mutations between these samples. Interestingly, we identified more predicted FSDNs in CrADs than in ECs, a recurrent finding that we also see in the analyzed HNPCC-Sys data, likely due to the immune pressure exerted on cancers. This pressure may eliminate clones with high mutational loads and FSDN expression, favoring those capable of immune evasion. Shared mutations and distinct differences between these samples were further validated in CRC and EC tumor cell lines, where shared mutations were also evident. This is consistent with our in vitro results, where we demonstrated that both in adenomas and healthy colorectal mucosa of LS previvors and survivors, there exist tissue-infiltrating T cells that can recognize our prioritized neoantigens, indicating that they are presented very early and can be recognized as pathogenic and form long-term memory responses. Interestingly, Bohaumilitzky et al observed dense CD8+T cell infiltration in the normal mucosa of patients with LS, indicating early recognition of FSDNs.17 This finding supports our focus on targeting FSDN in precursor lesions to enhance immunoprevention strategies in LS.
Our findings suggest that carriers of MLH1 and MSH2 pathogenic variants exhibit a higher frequency of FSDN-associated mutations compared with MSH6 carriers, aligning with established data that MLH1 and MSH2 mutations are associated with a greater mutation burden and higher rates of neoplastic occurrences.6 50 51 The differential in mutation load is significant regarding FSDN-based vaccines as it may directly impact their efficacy: MLH1 and MSH2 pathogenic variant carriers may present a more diverse and abundant pool of immunogenic targets, potentially leading to a stronger and more sustained vaccine-induced immune response. Conversely, MSH6 carriers, with fewer FSDNs, could have a less robust response to the same vaccine formulations. Therefore, tailored approaches based on the specific MMR mutation may be required to optimize vaccine efficacy.
Several clinical trials are currently exploring cancer vaccines based on FSDN as preventive strategies for LS. One of the first attempts to leverage FSDN vaccination in LS was made by Kloor et al, who conducted a Phase I/IIa clinical trial in 22 individuals with a history of stage III/IV MMRd CRC, employing a peptide vaccine targeting three FSDN (TAF1B, HT001, and AIM2).19 Vaccination was well tolerated and consistently induced both humoral and cellular immune responses; however, the CD8+T cell response was very limited. Another seminal study led by de Vries and colleagues at Radboud University Medical Center in Nijmegen investigated a DC-based vaccine targeting two FSDNs (TGFBR2 and CASP-5; NCT01885702). Unpublished data demonstrated strong T-cell responses against the TGFBR2 peptide with preliminary data suggesting a potential long-term decrease in cancer incidence among LS carriers who responded to this FSDN. In addition, NOUSCOM, a biotechnology company, is conducting a Phase 1b clinical trial (NCT05078866) evaluating the efficacy of an off-the-shelf viral vector-based cancer vaccine called NOUS-209 in LS carriers that encodes 209 shared FSDNs common in LS-associated tumors.12 Preliminary results from the first 10 LS individuals showed that the vaccine was safe, well-tolerated, and capable of generating potent and broad immunogenic T-cell responses.52 It is noteworthy that while several FSDNs in the referenced studies overlap with ours, we have focused on specific epitopes with varying affinities rather than the entire mutated protein. Although the full protein may coincide, the epitopes identified in our study are distinct. In this context, our group is initiating a Phase Ib study using autologous DCs loaded with pFSDNs. This approach aims to further refine immune-targeting strategies and contribute to the growing efforts in immunoprevention for LS.
We acknowledge several limitations in our study. First, although low-affinity (IC50<5000 nM) HLA binding predictions may raise concerns, we believe weak binders are key to preventing immune evasion by boosting recognition of overlooked antigens. We also demonstrated immune responses against these FSDNs. Second, while HLA class II predictions are less robust due to the promiscuous nature of these molecules, the tools we used remain the best available for this purpose. The inclusion of class II epitopes is crucial for eliciting CD4+T cell responses, which are vital in supporting long-lasting antitumor immunity. Third, our in vitro validation was conducted on a small cohort of patients. Despite this, the immune responses observed in these individuals suggest the potential efficacy of our approach and highlight the need for larger studies to validate these preliminary findings. We recognize that the limited cohort of sequenced LS samples in our study (comprising 17 CrADs and 8 ECs) constrains the generalizability of our findings. Nevertheless, the overlap of the presence of FSDN-associated mutations is clinically significant. For intellectual property reasons, we have not disclosed specific neoantigen sequences, which may limit external verification. While necessary to protect our work, we aim to balance transparency and commercial viability in future collaborations. Finally, we acknowledge that further validation through cytotoxicity assays is necessary to confirm the tumor-killing capacity of neoantigen-specific T cells, and these experiments are planned as part of our ongoing clinical trial.
In conclusion, this study presents a novel approach for identifying and validating FSDN that cover a wider population range and can serve as the basis for a universal cancer prevention vaccine development, as these peptides are expected to have broad potential in inducing immune memory to promote cancer prevention in LS carriers. A clinical trial to assess the safety and efficacy of a DC-based vaccine loaded with the pFSDN in generating neoantigen-specific immune responses is planned for early 2025.
Data availability statement
Data are available in a public, open access repository. Data available on: Bayó Llorens, Cristina (2024). Supplementary information. figshare. Dataset. https://doi.org/10.6084/m9.figshare.27969414.v3 Data and study materials will be made available upon request to the corresponding authors. Public data from the study HNPCC-Sys: Molecular Characterization of Lynch Syndromes can be accessed under request on dbGaP repository accession number phs001407.v1.p1. Supplementary tables available under Figshare:53 Bayó Llorens, Cristina (2024).
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the HCB Institutional Review Board (HCB/2019/0213), and all patients signed informed consent.
References
Footnotes
Contributors CB (conceptualization, investigation and analysis, writing original draft, visualization), GC (bioinformatic data analysis, writing original draft), FM (bioinformatic analysis), LM, JC-I, TO, HK, MP, LR, MD-A, OO, SC, RM (patient recruitment and clinical management), JC-H (sample analysis), MP and GF-G (supervision, review and editing), MJ (supervision, resources), DB-R and FB (conceptualization, funding acquisition, supervision, review and editing). FB is the guarantor for this study.
Funding This study was funded by the Instituto de Salud Carlos III (PI19/01867; PI22/00470; ICI22/00063), and co-funded by the European Union, and through a Retos Investigación grant (PID2019-111254RB-I00 and PID2023-151585OB-I00). CIBERehd and CIBERONC (CB16/12/00234) is funded by the Instituto de Salud Carlos III. Also was funded by Projecte 2023_LLAV_00111, with the support from Departament de Recerca i Universitats de la Generalitat de Catalunya (AGAUR, grant 2023SGR01112; PERIS_SuportGrups23, SLT028/23/000212). JC-I has been funded by Contractes Clínic de Recerca "Emili Letang - Josep Font" 2022, granted by Hospital Clínic de Barcelona.
Competing interests FB received consultant fees from Olympus, Persei Vivarium, Biodexa, Nouscom, Sysmex, and Nor-gine, and editorial fees from Elsevier.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.