Article Text

Abstract
Background The stimulator of interferon genes (STING) signaling pathway has been demonstrated to propagate the cancer-immunity cycle and remodel the tumor microenvironment and has emerged as an appealing target for cancer immunotherapy. Interest in STING agonist development has increased, and the candidates hold significant promise; however, most are still in the early stages of human clinical trials. We found that ABT-199 activated the STING pathway to enhance the immunotherapeutic effect, and provided a ready-to-use small molecule drug for STING signaling activation.
Methods Phosphorylation of STING, TBK1, and IRF3, as well as activation of the interferon-I (IFN-I) signaling pathway, were detected following ABT-199 treatment in various colorectal cancer cells. C57BL/6J and BALB/c mice with subcutaneous tumors were employed to evaluate the in vivo therapeutic effects of the ABT-199 and anti-PD-L1 combination. Flow cytometry and ELISA were employed to analyze the level and activity of tumor-infiltrating T lymphocytes. Immunofluorescence and quantitative real-time PCR were conducted to assess the source and accumulation of double stranded DNA (dsDNA) in the cytoplasm. Chemical cross-linking assay, co-immunoprecipitation, and CRISPR/Cas9-mediated knockout were performed to investigate the molecular mechanism underlying ABT-199-induced voltage-dependent anion channel protein 1 (VDAC1) oligomerization and mitochondrial DNA (mtDNA) release.
Results ABT-199 significantly activated the STING signaling pathway in various colorectal cancer cells, which was evidenced by increased phosphorylation of TBK1 and IRF3, and upregulation of C-C motif chemokine ligand 5 (CCL5), C-X-C motif chemokine ligand 10 (CXCL10), and interferon beta transcription. By promoting chemokine expression and cytotoxic T-cell infiltration, ABT-199 promoted antitumor immunity and synergized with anti-PD-L1 therapy to improve antitumor efficacy. ABT-199 induced mtDNA accumulation in the cytoplasm and triggered STING signaling via the canonical pathway. cGAS or STING-KO models significantly abolished both STING signaling activation and the antitumor efficacy of ABT-199. Mechanically, ABT-199 promoted VDAC1 oligomerization by disturbing the binding between BCL-2 and VDAC1, thereby facilitating mtDNA release into the cytoplasm. ABT-199-triggered STING signaling was attenuated when VADC1 was knocked out. Consistently, the antitumor effect of ABT-199 in vivo was abolished in the absence of VDAC1.
Conclusions Our results identify a ready-to-use small molecule compound for STING activation, reveal the underlying molecular mechanism through which ABT-199 activates the STING signaling pathway, and provide a theoretical basis for the use of ABT-199 in cancer immunotherapy.
- Colorectal Cancer
- Immunotherapy
- Cytokine
- Immune Checkpoint Inhibitor
Data availability statement
Data are available upon reasonable request. No data are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Previous studies have demonstrated that activation of the stimulator of interferon gene (STING) signaling pathway enhances both innate and adaptive immunity in cancer and serves as an effective strategy to potentiate the efficacy of immune checkpoint inhibitors. Although there has been increased development of STING agonists, no clinically available small molecule drug has yet been identified for activating STING signaling to enhance the therapeutic outcomes of immunotherapy in colorectal cancer.
WHAT THIS STUDY ADDS
Our study demonstrates that ABT-199 efficiently activates the STING signaling pathway, modulates the interferon-I signaling pathway, and upregulates the expression of T cell-related chemokines in tumor tissues. Moreover, ABT-199 significantly improves the antitumor efficacy of anti-PD-L1 therapy. At the mechanistic level, ABT-199 promotes voltage-dependent anion channel protein 1 (VDAC1) oligomerization by disturbing the binding between BCL-2 and VDAC1, resulting in the release of mitochondrial DNA (mtDNA) into the cytoplasm.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study identifies ABT-199 as a new small-molecule drug for STING activation, elucidates the underlying molecular mechanisms of BCL-2-mediated STING activation through mtDNA, and offers valuable insights and a potential strategy to enhance the efficacy of immunotherapy in colorectal cancer.
Background
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon gene (STING) signaling pathway, which senses cytosolic DNA to trigger type I interferon (IFN-I) signaling, has recently been demonstrated to activate both innate and adaptive immunity in cancer.1–3 STING activation contributes to cancer cell death and the induction of T-cell responses. Through the elicitation of IFN-I secretion, STING signaling facilitates dendritic cell maturation, thereby enhancing antigen processing and presentation. STING activation promotes the priming and activation of T cells by enhancing signaling, including tumor antigens presented on major histocompatibility complexes (signal 1), costimulatory molecules (signal 2), and certain proinflammatory cytokines (signal 3). STING pathway activation also contributes to the trafficking and infiltration of T cells into tumors by both increasing the expression of T-cell chemokines and normalizing the tumor vasculature.4–6 Furthermore, STING activation can upregulate major histocompatibility complex class I molecules on cancer cells and improve tumor antigen presentation, thus promoting their recognition and elimination by cytotoxic T cells.7
The immunomodulatory roles of STING enable it to be an appealing target for cancer immunotherapy, and interest in developing STING agonists has increased.5 8 Cyclic dinucleotides (CDNs) are natural agonists of STING, which have been proven to have good antitumor effects and representative molecules, have advanced into clinical trials.9 However, the chemical properties of CDNs, such as poor metabolic stability, poor tissue permeability and tumor targeting specificity greatly hinder their clinical application.10 Therefore, the focus of STING agonist development has shifted to non-CDN-derived small molecules, which usually have superior pharmacokinetic properties and can achieve more accurate dose control and systemic administration. Further, the structure is easier to modify and optimize, which allows researchers to fine-tune their structure to improve drug specificity and reduce potential side effects. By designing and optimizing non-CDN STING agonists, the precise regulation of the STING signaling pathway can be achieved, the incidence of non-specific inflammatory reactions can be reduced, and the safety and efficacy of drugs can be improved.11 Although these candidates show strong promise, most of them are still in phase I of clinical trials and have a long way to go.3 8 In academia, PARP inhibitors have been proven to promote the accumulation of cytosolic DNA fragments and activate the cGAS-STING pathway, thereby stimulating the production of IFN-I to induce antitumor immunity.12 13 Cytotoxic agents, including topoisomerase inhibitors and cisplatin, activate the STING signaling pathway to enhance CD8+ T-cell infiltration and synergize with immune checkpoint inhibitors (ICIs) to achieve tumor regression.14–16 These findings provide new insight into the triggering of the STING pathway and a ready-to-use small molecule drug for STING signaling activation.
In this study, we found that the selective BCL-2 inhibitor ABT-199 activated the STING signaling pathway and enhanced CXCL10 and interferon beta (IFNβ) secretion in colorectal cancer (CRC) cells. Compared with the STING agonists in clinical trials, ABT-199, which has been approved for the treatment of acute myelogenous leukemia, could be a ready-to-use small molecule drug for STING signaling activation. ABT-199 promoted cytotoxic T-cell infiltration, exerted antitumor immunity, and improved the therapeutic efficacy of programmed death-ligand 1 (PD-L1) antibodies. ABT-199 caused mitochondrial-derived double stranded DNA (mtDNA) accumulation in the cytoplasm and subsequently triggered the STING signaling pathway. Mechanistically, BCL-2 physically interacted with voltage-dependent anion channel protein 1 (VDAC1), whereas ABT-199 disrupted this interaction to enable VDAC1 oligomerization; this, in turn, led to the release of mtDNA and the activation of the STING signaling pathway. In vivo, both cGas, Sting, and Vdac1 depletion abolished the antitumor effects of ABT-199. Overall, ABT-199 has the potential to activate the STING signaling pathway to augment the antitumor effects of ICIs in CRC.
Materials and methods
Western blot analysis
After the indicated treatments, cells were harvested and boiled for 15 min in 2.5×sodium dodecyl sulfate (SDS) loading buffer. The proteins were then resolved by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% non-fat dry milk for 1 hour at room temperature and incubated with indicated primary antibodies at 4°C overnight. After washes with phosphate-buffered saline (PBS), the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody in non-fat dry milk (1:5,000) for 1 hour at room temperature. After washes with phosphate buffered saline with Tween (PBST), the membranes were exposed and detected by Amersham Imager 600 (GE HealthCare, Little Chalfont, Buckinghamshire, UK).
RNA isolation and quantitative real-time PCR
Total RNA of all harvested samples was extracted with TRIzol reagent (#9109, Takara, Tokyo, Japan) and quantified using the NanoDrop Spectrophotometer ND-1000 (Thermo Fisher Scientific, Waltham, Massachusetts, USA). 2 µg total RNA was used for complementary DNA (cDNA) synthesis using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (AT311-03, TransGen Biotech, Beijing, China). Quantitative real-time PCR was performed with the SYBR Green kit (#172–5124, Bio-Rad, Hercules, California, USA) in a CFX96 Touch Real-Time PCR Instrument (Bio-Rad, Hercules, California, USA). Fold changes in gene expression were calculated using a comparative threshold cycle (Ct) method, with β-Actin as the internal control. Quantitative real-time PCR (qPCR) experiments were performed with technical replicates, setting up replicate wells at the same time, ensuring that the difference in Ct values was <0.5 to guarantee reproducibility. The primers used are listed in online supplemental table 2.
Supplemental material
Flow cytometry
Cell apoptosis detection. HT-29 and CT26 cells were treated with ABT-199 (5 µM) for the indicated time. Cell supernatants were collected and cells were obtained with pancreatin without EDTA. Following treatment, cells were washed with PBS and centrifuged for 4 min at 1,600 rpm. Apoptosis assays were performed using the Annexin V/PI Apoptosis Kit (AP101, MultiSciences Biotech) as follows: cell pellets were resuspended in 500 µL 1×Binding Buffer, and stained with Annexin V-FITC and PI for 5 min at room temperature (protected from light). For positive controls, cells were heated for 1 min and then cooled on ice, stained with Annexin V-FITC or PI for 5 min at room temperature. The cellular apoptotic rate was analyzed by CytoFLEX (Beckman Coulter, Miami, Florida, USA).
Mitochondrial membrane potential detection. HT-29 and CT26 cells were treated with ABT-199 (1.25, 2.5, and 5 µM) for 24 hours. The untreated cells were treated with 10 µM Carbonyl cyanide m-Chlorophenylhydrazone (CCCP) at 37°C for 20 minutes. Following treatment, collected cells and supernatants were washed with PBS and centrifuged for 4 min at 1,600 rpm. Mitochondrial membrane potential was measured using the JC-1 kit (C2006, Beyotime) as follows: cell pellets were resuspended in 500 µL 1×JC-1 Buffer at 37°C for 20 min and flipped every 5 min to prevent cell clustering. Mitochondrial membrane potential was detected by CytoFLEX (Beckman Coulter, Miami, Florida, USA).
The immune cell infiltration and membrane PD-L1 expression detection. Mouse cancer tissues were clipped to 5 mL protein extraction buffer (PEB) lysis buffer (2 mM EDTA, 0.5% bovine serum albumin (BSA)) and homogenized with tissue dissociators (Miltenyi Biotec, Bergisch Gladbach, Germany). After being washed with PBS, each sample, as well as the fluorescence minus one (FMO) control for each individual color, was incubated with Fc receptor-blocking solution for 10 min, and stained with the following fluorochrome-conjugated antibodies or an isotype: anti-mouse CD45 (#103106, BioLegend), anti-mouse CD3 (#100204, BioLegend), anti-mouse CD8a (#100705, BioLegend) at room temperature for 30 min, and with anti-mouse PD-L1 (#124307, BioLegend) at 4°C for 2 hours (1 µL/2×105 cells in 100 µL 0.2% BSA). 7-aminoactinomycin D-PerCP/Cy5.5 (#555816, BD Biosciences) was stained simultaneously for 10 min. After washes, samples were measured by CytoFLEX (Beckman Coulter, Miami, Florida, USA).
Mitochondria isolation
The intact mitochondria were isolated according to the protocol of the mitochondria isolation kit (Beyotime, C3601). Briefly, cells were plated in 10 cm dishes and treated with ABT-199 (5 µM) or DMSO alone for 24 hours. After treatment, cells were harvested and resuspended in the mitochondria extraction reagent (1 mL/2×107 cells). After standing in the ice bath for 10 min, the resuspended cells were homogenized about 10–30 times and centrifuged at 600 g at 4°C for 10 min. Supernatants were collected and centrifuged at 6,000 g for 10 min, and the final pellet was mitochondria.
Animal experiments
All animal experiments were performed following the guidelines approved by the Institute of Animal Care and Use Committee of Zhejiang University in Hangzhou (IACUC no.19–136, 19–262). Single cells were obtained by digestion of CT26, cGas-KO CT26, Sting-KO CT26, Vdac1-KO CT26, or MC38 cells and injected subcutaneously into 4–5-week-old male BALB/c or C57BL/6 mice (Beijing Vital River Laboratory Animal Technology, Beijing, China). EMT6 cells were injected subcutaneously into 5-week-old BALB/c female mice (GemPharmatech, Jiangsu, China). Mice were bred and maintained under specific pathogen-free conditions, and were provided sterilized food and water ad libitum at the Center for Drug Safety Evaluation and Research of Zhejiang University. Tumor volumes were measured on the day of administration and calculated every other day using the following formula: volume (mm3)=length×width×width/2. For treatment, mice were intravenously injected with 10 mg/kg anti-PD-L1 twice a week and dosed orally with 100 mg/kg ABT-199 daily. Mice were sacrificed and tumors were harvested for flow cytometry or immunoblot analysis.
Statistical analysis
All of the data are presented as mean±SD. Statistical significance of the differences was determined by Student’s t-test (two groups), one-way analysis of variance (ANOVA) with Dunnett’s post hoc test (more than two groups), or two-way ANOVA with Dunnett’s post hoc test (more than two groups with more than one factor). Statistical analysis was conducted with GraphPad Prism V.9 (GraphPad Software, La Jolla). n.s, not significant. For western blot, the densitometric analysis was performed using Image J (National Institutes of Health).
Results
ABT-199 results in cGAS-STING signaling pathway activation
Given that the activation of the STING signaling pathway leads to the upregulation of C-X-C motif chemokine ligand 10 (CXCL10) and IFNβ expression, we conducted an ELISA to systematically screen a panel of small-molecule compounds with the capacity to induce the activation of the STING pathway (online supplemental table 1). The CRC cell line HT-29, a representative microsatellite-stable (MSS) CRC cell line with cell-intrinsic STING expression and function, was used as a cell model for compound screening.17 18 We first performed the validation in HT-29 cells, and the results showed that the phosphorylation levels of STING, TBK1, and IRF3 were increased on treatment with the STING agonist htDNA, indicating that htDNA significantly activated the STING signaling pathway in HT-29 cells (online supplemental figure 1A). We used 5-fluorouridine, a compound previously reported to elicit the secretion of CXCL10 and IFNβ, as a positive control. There are 161 clinically used and tested anticancer drugs, and each drug in the library was added at a concentration of 5 µM. The selective BCL-2 inhibitor ABT-199 prominently increased the concentrations of CXCL10 and IFNβ in the cell culture medium (figure 1A). Furthermore, ABT-199 obviously upregulated the messenger RNA (mRNA) levels of C-C motif chemokine ligand 5 (CCL5), CXCL10, and IFNβ in a dose and time-dependent manner (figure 1B and C), and the mRNA levels were increased at 24 hours and peaked at 72 hours post-treatment. To ascertain whether this elevation stemmed from STING signaling activation, we assessed STING signaling. ABT-199 significantly induced STING, TBK1, and IRF3 phosphorylation (figure 1D and E). This trend was consistent across various microsatellite instability (MSI) and MSS CRC cell lines, including SW480, SW620, SW48, and RKO cells (figure 1F). Similarly, ABT-199-induced STING, TBK1, and IRF3 phosphorylation was verified in primary cancer cells derived from patients with CRC (online supplemental figure 1B) . These collective findings conclusively established the capacity of ABT-199 to activate the STING signaling pathway in CRC.

ABT-199 activated the STING signaling pathway in colorectal cancer. (A) The effect of small-molecule drugs on CXCL10 and IFNβ secretion. HT-29 cells were treated with a compound library containing 161 clinically used and tested anticancer drugs, at a concentration of 5 µM for 24 hours, then cell-free supernatants were collected and analyzed for CXCL10 and IFNβ secretion by ELISA. Relative expression compared with the control group was shown with a heatmap. Control group: HT-29 cells were treated with DMSO alone. (B and C) Transcription levels of CCL5, CXCL10, and IFNβ in HT-29 cells treated with ABT-199 (1.25, 2.5, and 5 µM) for 24 hours (B) or cells treated with ABT-199 (5 µM) for 24, 48, and 72 hours (C) were analyzed by qRT-PCR. (D and E) The P-TBK1, TBK1, P-IRF3, IRF3, P-STING, and STING proteins in HT-29 cells with indicated concentrations of ABT-199 (1.25, 2.5, and 5 µM) for 24 hours (D) or cells treated with ABT-199 (5 µM) for 24, 36, and 48 hours (E) were evaluated by western blot. The relative expression of P-TBK1, P-IRF3, and P-STING was analyzed quantitatively by Image J analysis. (F) The P-TBK1, TBK1, P-STING, and STING proteins in MSS cell lines (SW480 and SW620) and MSI cell lines (SW48 and RKO) treated with ABT-199 (5 µM) for 24 hours were evaluated by western blot. Data represent the mean±SD (C and E, n=3; B and D, n=4). The statistical difference in the data was analyzed by one-way analysis of variance with Dunnett’s post hoc test. CCL5, C-C motif chemokine ligand 5; CXCL10, C-X-C motif chemokine ligand 10; DMSO, dimethyl sulfoxide; IFNβ, interferon beta; MSI, microsatellite instability; MSS, microsatellite-stable; mRNA, messenger RNA; n.s, no significance; qRT-PCR, quantitative reverse transcription polymerase chain reaction; STING, stimulator of interferon genes.
Next, we investigated whether ABT-199 could activate the STING signaling pathway in murine cancer cells due to species-specific differences between murine Sting and human STING.5 We used the mouse CRC cell lines CT26 and MC38, which harbor MSS and MSI, respectively, thus mirroring the true clinical status of patients.19 The data demonstrated that ABT-199 effectively induced the phosphorylation of STING and TBK1 in CT26 and MC38 cells (online supplemental figure 1C,D). Concurrently, ABT-199 elicited the mRNA expression of Ccl5, Cxcl10, and Ifnβ (online supplemental figure 1E). Notably, this phenomenon was also evident in the breast cancer cell line EMT6 (online supplemental figure 1F). Taken together, these data indicated that ABT-199 activated both human and murine STING signaling, which facilitated our subsequent in vivo study.
ABT-199 increases the efficacy of immune checkpoint blockade
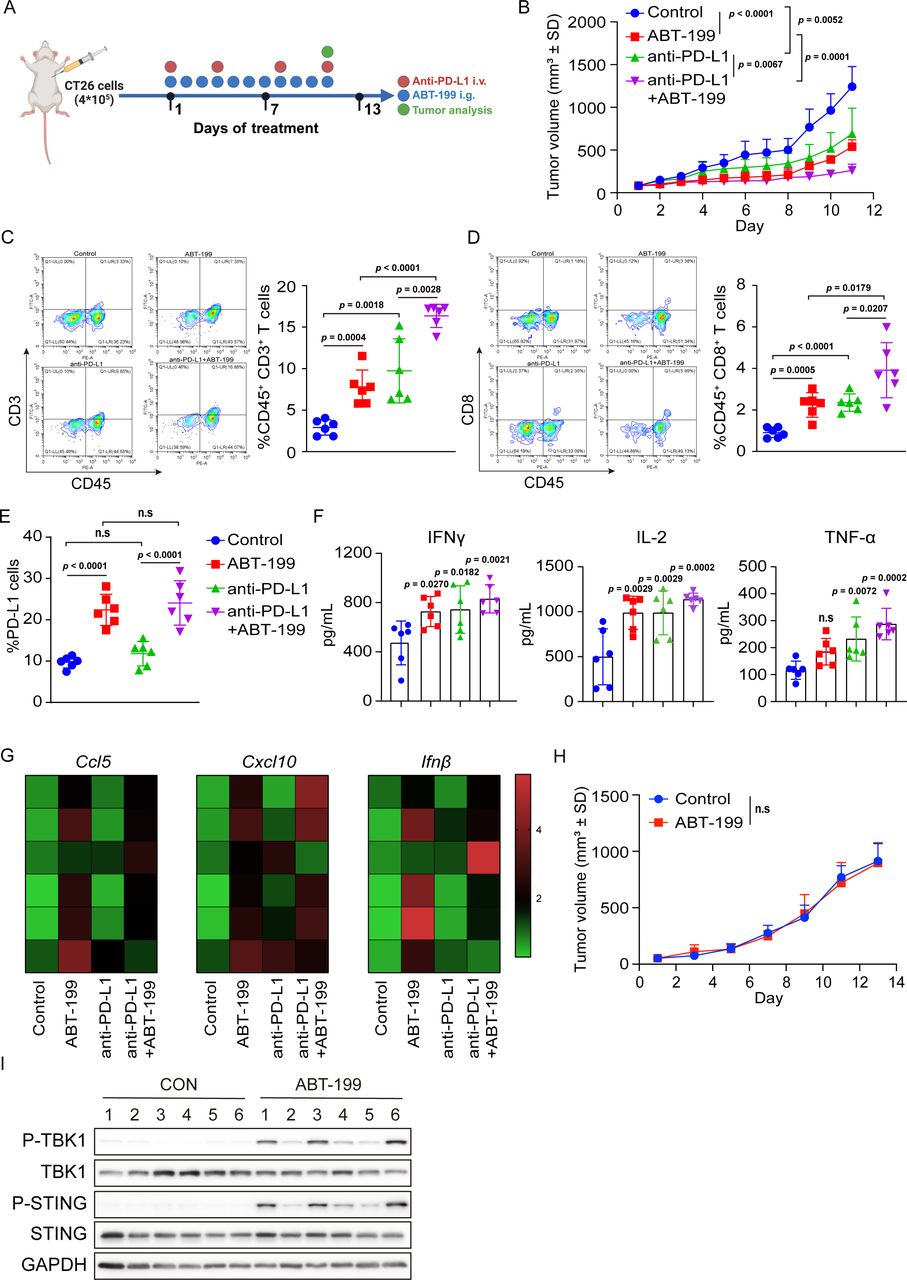
Subsequently, we sought to determine whether ABT-199 could activate STING in vivo and alter the tumor microenvironment. We used a CT26 mouse cancer model, which harbors MSS and partially responds to anti-programmed cell death protein 1 (PD-1)/PD-L1 therapy. The mice were treated with ABT-199 or anti-PD-L1 antibody as indicated (figure 2A). Notably, ABT-199 significantly inhibited tumor growth, and compared with ABT-199 monotherapy, ABT-199 combined with anti-PD-L1 synergistically exerted antitumor effect (figure 2B). In tumors, ABT-199 increased the infiltration of CD3+ and CD8+ T cells, while this effect was further enhanced in the ABT-199 plus anti-PD-L1 group (figure 2C and D). Additionally, ABT-199 upregulated membranous PD-L1 expression in both the ABT-199 group and the combination group (figure 2E). This intervention also triggered increased secretion of IFN-γ, interleukin (IL)-2, and tumor necrosis factor (TNF)-α, as evidenced by ELISA analysis (figure 2F). Quantitative reverse transcription polymerase chain reactionq (RT-PCR) analysis of tumor tissues revealed that ABT-199 induced the mRNA expression of Ccl5, Cxcl10, and Ifnβ (figure 2G). To determine whether ABT-199-mediated tumor regression was dependent mainly on the immune system, we conducted a parallel investigation in immunodeficient BALB/c nude mice. As a result, no significant antitumor effect was observed in the absence of the immune system (figure 2H), despite ABT-199 inducing TBK1 and STING phosphorylation within the tumors (figure 2I). These findings collectively suggested that ABT-199 might exert antitumor effects in vivo by activating the STING signaling pathway.

ABT-199 synergized with anti-PD-L1 to inhibit CT26 tumor growth in vivo. (A) Schematic diagram of the experimental procedure: BALB/c mice were inoculated with CT26 cells. Subsequently, the mice were treated with ABT-199 (100 mg/kg) and anti-PD-L1 (10 mg/kg) either individually or in combination. Tumor volume, T-cell infiltration, and the STING signaling pathway were subsequently assessed. (B) Tumor growth curves of immunocompetent BALB/c mice treated with ABT-199, anti-PD-L1, or a combination of ABT-199 and anti-PD-L1. (C and D) The tumor-infiltrating CD45+CD3+ T cells (C) and CD45+CD8+ T cells (D) were detected by flow cytometry. (E) Cell-surface PD-L1 was analyzed by flow cytometry. (F) Tumor-secreted IFNγ, IL-2, and TNF-α were collected and analyzed by ELISA. (G) Tumor tissues were separated and the messenger RNA levels of Ccl5, Cxcl10, and Ifnβ were measured by qRT-PCR. (H) Tumor growth curves of BALB/c nude mice treated with ABT-199 (100 mg/kg). (I) CT26 tumor tissues were separated and the P-TBK1, P-STING, TBK1, and STING expression were detected by western blot. Data were presented as the mean±SD (n=6). Statistical analysis of the data was performed by Student’s t-test (two groups) and one-way analysis of variance with Dunnett’s post hoc test (more than two groups). CCL5, C-C motif chemokine ligand 5; CXCL10, C-X-C motif chemokine ligand 10; IFN, interferon; i.g., intragastric; IL, interleukin; i.v., intravenous injection; n.s, no significance; PD-L1, programmed death-ligand 1; STING, stimulator of interferon gene; TNF, tumor necrosis factor.
We analyzed the antitumor immune response elicited by ABT-199 in the MC38 mouse cancer model, which is characterized by MSI. Notably, the administration of ABT-199 significantly delayed tumor growth and potentiated the effects of anti-PD-L1 therapy (online supplemental figure 2A) without substantially altering body weight (online supplemental figure 2B). Furthermore, cotreatment with ABT-199 and anti-PD-L1 antibody further increased CD3+ and CD8+ T-cell infiltration relative to that induced by anti-PD-L1 antibody (online supplemental figure 2C,D). In parallel, ABT-199 triggered the STING signaling pathway within the tumors (online supplemental figure 2E).
To ascertain the generalizability of the synergy between ABT-199 and anti-PD-L1, we evaluated the therapeutic impact of ABT-199 and anti-PD-L1 in EMT6 tumor-bearing mice. The mice with established tumors were treated with ABT-199 and anti-PD-L1 antibody alone or in combination (online supplemental figure 3A). The results showed that the combination of ABT-199 and anti-PD-L1 antibody resulted in sustained control of tumor growth (online supplemental figure 3B). This combined treatment also substantially augmented the infiltration of intratumoral CD3+ and CD8+ T cells (online supplemental figure 3C,D). Similarly, ABT-199 treatment elicits an elevation in the expression of PD-L1 on the cell membrane (online supplemental figure 3E). Moreover, increases in IFN-γ, IL-2, and TNF-α were observed with ABT-199 and anti-PD-L1 treatment alone or in combination (online supplemental figure 3F). ABT-199 also had similar effects on activating the STING signaling pathway (online supplemental figure 3G). Together, these data indicated that the activation of the STING signaling pathway by ABT-199 synergistically cooperated with ICIs, thereby culminating in effective tumor suppression.
ABT-199 activates the STING signaling pathway by inducing mtDNA release
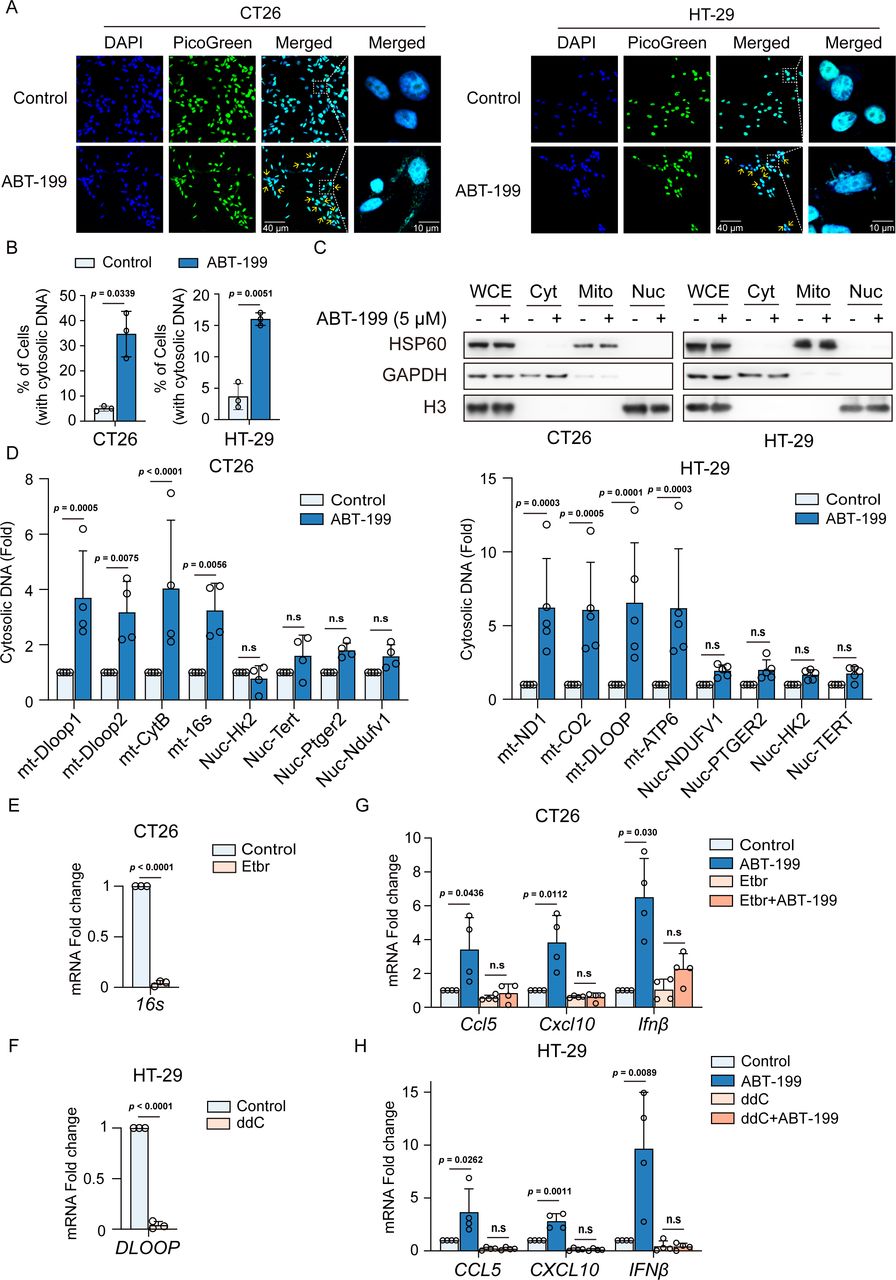
We proceeded to explore the mechanism underlying the activation of the STING pathway by ABT-199. As an innate cytosolic DNA-sensing pathway, STING signaling is directly initiated by dsDNA within the cytoplasm.20 Hence, our initial focus was to determine whether ABT-199 promoted the accumulation of cytoplasmic dsDNA. Consequently, ABT-199 treatment led to a significant increase in cytosolic dsDNA in CT26 and HT-29 cells (figure 3A and B). Since liberated dsDNA primarily emanates from nuclear DNA (nDNA) and mtDNA, we endeavored to pinpoint the source of cytosolic DNA. There was a significant difference in the mtDNA and nDNA genomes, which could be classified by mtDNA-specific or nDNA-specific primers. We fractionated cell extracts into cytosolic, mitochondrial, and nuclear fractions and detected GAPDH, HSP60, and histone H3 in the respective fractions (figure 3C). Subsequently, we purified DNA from each fraction and quantified DNA containing specific mitochondrial genes (DLOOP, CYTB, 16S, ND1, CO2, and ATP6) and nuclear genes (HK2, TERT, PTGER2, and NDUFV) using qPCR. ABT-199-treated cells exhibited a substantial increase in mtDNA within the cytosolic fraction (figure 3D). To determine whether the release of mtDNA was responsible for the activation of the STING pathway, we generated mtDNA-deficient cells using ethidium bromide (EtBr) and dideoxycytidine (ddC).21 22 As anticipated, prolonged EtBr or ddC treatment resulted in substantial depletion of mtDNA (figure 3E and F). Additionally, the absence of mtDNA diminished ABT-199-induced CCL5, CXCL10, and IFNβ mRNA expression (figure 3G and H). Furthermore, mtDNA depletion induced by EtBr treatment also abrogated ABT-199-induced phosphorylation of TBK1 and STING (online supplemental figure 4A).

ABT-199 induced mitochondrial-derived cytosolic dsDNA release to activate the stimulator of interferon gene signaling pathway. (A and B) Representative images (A) and quantitative analysis (B) of dsDNA reagent staining in CT26 and HT29 cells treated with DMSO or ABT-199 (5 µM) for 24 hours. DAPI (blue) was used to visualize the nucleus. Scale bar: 40 µm and 10 µm. (C) CT26 and HT29 cells were treated with ABT-199 (5 µM) for 24 hours, then fractionated into WCE, Cyt, Mito, and Nuc fractions and subjected to analysis using western blot. GAPDH, HSP60, and H3 were used as markers for Cyt, Mito, and Nuc fractions. (D) DNA from each fraction was purified, and Dloop1, Dloop2, CytB, 16s, Hk2, Tert, Ptger2, and Ndufv1 expression levels were detected by qRT-PCR. (E) CT26 cells were treated with EtBr (250 ng/mL) for 7 days, and the mRNA levels of 16s were evaluated by qRT-PCR. (F) HT-29 cells were treated with ddC (1 µM) for 7 days, and the mRNA levels of DLOOP were evaluated by qRT-PCR. (G) CT26 cells were treated with EtBr (250 ng/mL) for 7 days, and then treated with ABT-199 (5 µM) for 24 hours. Subsequently, transcription levels of Ccl5, Cxcl10, and Ifnβ mRNA were quantified by qRT-PCR. (H) HT-29 cells were treated with ddC (1 µM) for 7 days, followed by ABT-199 (5 µM) treatment for 24 hours. Then transcription levels of CCL5, CXCL10, and IFNβ mRNA were quantified using qRT-PCR. Data represented the mean±SD (B, E and F, n=3; D, G and H, n=4). Statistical analysis of the data was performed by Student’s t-test (two groups) and two-way analysis of variance with Dunnett’s post hoc test (more than two groups). CCL5, C-C motif chemokine ligand 5; CXCL10, C-X-C motif chemokine ligand 10; DAPI, 4',6-Diamidino-2-phenylindole; ddC, dideoxycytidine; DMSO, dimethyl sulfoxide; double stranded DNA, dsDNA; EtBr, ethidium bromide; IFN, interferon; mRNA, messenger RNA; n.s, no significance; qRT-PCR, quantitative reverse transcription polymerase chain reaction.
Recent studies have revealed that ABT-199 facilitates dendritic cells (DC)-mediated antigen presentation by activating the mtDNA-cGAS-STING pathway,23 and we further addressed this experimentally in our system. We detected the cGAS-STING pathway activation in the mouse dendritic cell line DC2.4 and mouse bone marrow dendritic cells (BMDCs) after ABT-199 treatment. The results showed that ABT-199 promoted the phosphorylation levels of STING and TBK1 in both DC2.4 cells and BMDCs (online supplemental figure 4B). In addition, DC2.4 cells were fractionated into cytosolic, mitochondrial, and nuclear fractions, and qPCR results showed that ABT-199 administration led to the accumulation of mitochondrial DNA within the cytosolic fraction (online supplemental figure 4C,D), which was consistent with our findings in tumor. We next examined whether the release of mtDNA induced by ABT-199 was associated with mitochondrial damage. Alterations in the mitochondrial membrane potential, an early hallmark of mitochondrial damage, were assessed using a mitochondrial membrane potential assay kit with JC-1 to evaluate mitochondrial damage.24 Remarkably, CT26 and HT-29 cells exhibited a reduction in the mitochondrial membrane potential following ABT-199 treatment (online supplemental figure 4E). Collectively, these results showed that ABT-199 induced mtDNA release to the cytosol, thereby activating the STING signaling pathway.
Intratumoral STING pathway activation is required for ABT-199-induced antitumor efficacy
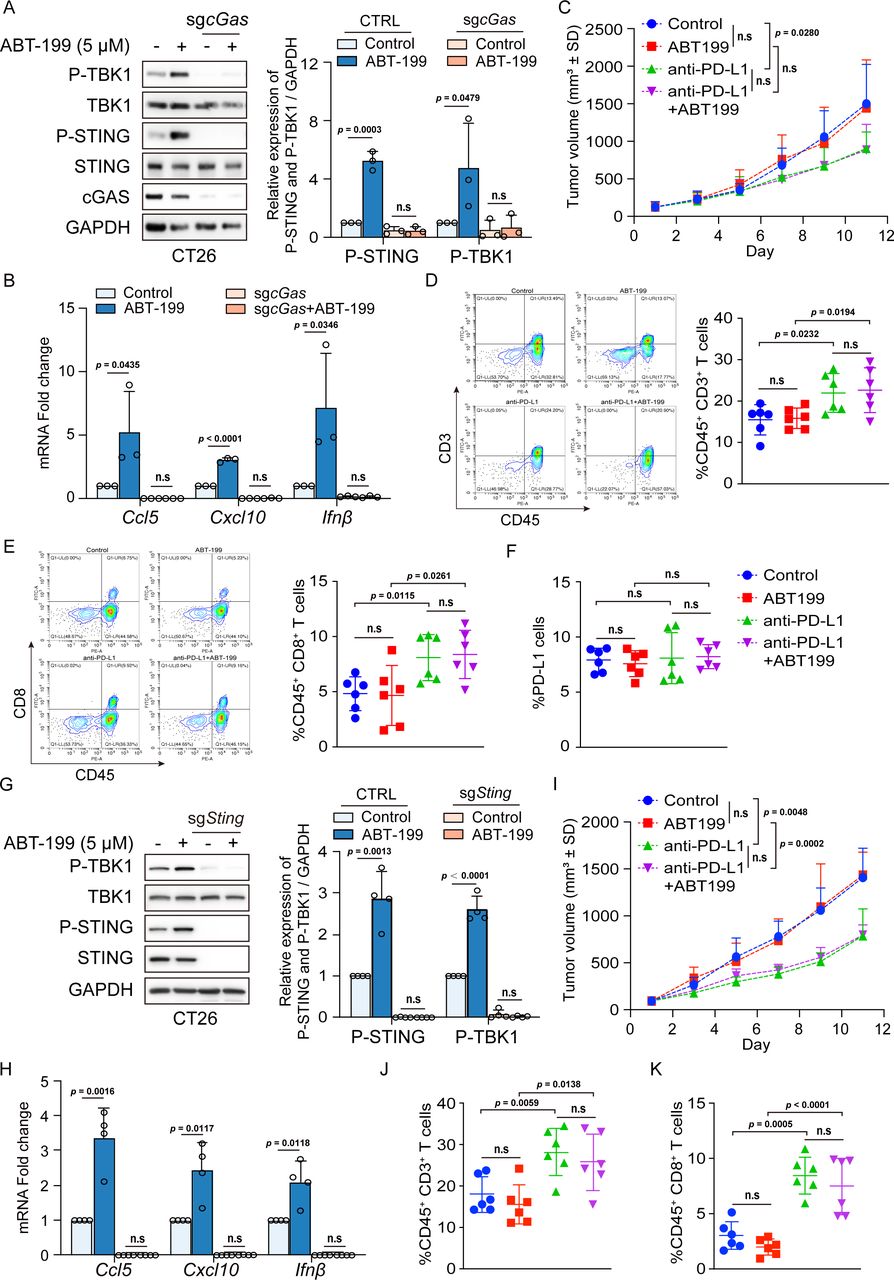
To determine whether the DNA sensor cGAS is required for cytokine production and T-cell recruitment in response to ABT-199, we employed CRISPR/Cas9 to knock out cGas for in-depth examination. As shown in figure 4A, ABT-199-induced TBK1 and STING phosphorylation was abolished following cGas knockout. Consequently, the changes in the mRNA levels of Ccl5, Cxcl10, and Ifnβ in response to ABT-199 were eliminated in cGas-knockout cells (figure 4B). Furthermore, we assessed whether the STING signaling pathway was required for ABT-199-mediated tumor regression in vivo. BALB/c mice were subcutaneously inoculated with cGas-knockout CT26 cells, and then, the mice were administered with ABT-199 and anti-PD-L1 antibody alone or in combination treatment. The results showed that ABT-199 could not inhibit tumor growth and did not show synergy with anti-PD-L1 therapy to enhance antitumor efficacy (figure 4C). Similarly, no significant difference in tumor infiltration by CD3+ or CD8+ T cells on ABT-199 treatment was detected (figure 4D and E). Similarly, cGAS deficiency significantly attenuated ABT-199-induced PD-L1 expression (figure 4F). To further validate that ABT-199-induced downstream effects are specifically mediated by STING, we also depleted Sting in CT26 cells. As observed with cGas depletion, Sting depletion completely impaired ABT-199-induced phosphorylation of TBK1 and STING (figure 4G) and increased in mRNA levels of Ccl5, Cxcl10, and Ifnβ (figure 4H). Additionally, we examined the effect of ABT-199 antitumor and its synergy with anti-PD-L1 antibody in Sting-KO mouse models, and the results showed that the inhibition of tumor growth by ABT-199 was significantly reversed in Sting-KO tumors. The synergistic effect of ABT-199 and anti-PD-L1 antibody was also abolished compared with the anti-PD-L1 group (figure 4I). Importantly, the ABT-199-induced increase in both tumor-infiltrating CD3+ and CD8+ T cells was also reduced on Sting depletion (figure 4J and K). These data indicated that the STING pathway plays a critical role in ABT-199-induced T-cell recruitment and tumor regression.

cGAS or STING depletion impaired ABT-199-induced proinflammatory signaling and antitumor efficacy. (A and B) CRISPR/Cas9 control and cGas-KO cells were administered with ABT-199 (5 µM) for 24 hours and subjected to immunoblotting for P-TBK1, TBK1, P-STING, STING, and cGAS levels (A), or qRT-PCR for Ccl5, Cxcl10 and Ifnβ mRNA (B). (C) The tumor volume of BALB/c mice bearing the cGas-KO CT26 cells after treatment of ABT-199 (100 mg/kg) and anti-PD-L1 (10 mg/kg) alone or in combination. (D and E) The infiltration of CD45+CD3+ T cells (D) and CD45+CD8+ T cells (E) into tumors was measured by flow cytometry. (F) PD-L1 levels on the surface of tumor cells were analyzed by flow cytometry. (G and H) CRISPR/Cas9 control and Sting-KO cells were administered with ABT-199 (5 µM) for 24 hours and subjected to immunoblotting for P-TBK1, TBK1, P-STING, and STING levels (G), or qRT-PCR for Ccl5, Cxcl10 and Ifnβ mRNA (H). (I) The tumor volume of BALB/c mice bearing the Sting-KO CT26 cells after treatment of ABT-199 (100 mg/kg) and anti-PD-L1 (10 mg/kg) alone or in combination. (J and K) The infiltration of CD45+CD3+ T cells (J) and CD45+CD8+ T cells (K) into tumors was measured by flow cytometry. Data were presented as the mean±SD (A and B, n=3; G and H, n=4; C, D, E, F, I, G, and K, n=6). Statistical analysis of the data was performed by Student’s t-test (two groups). CCL5, C-C motif chemokine ligand 5; cGAS, cyclic GMP-AMP synthase; CXCL10, C-X-C motif chemokine ligand 10; IFN, interferon; mRNA, messenger RNA; n.s, no significance; PD-L1, programmed death-ligand 1; qRT-PCR, quantitative reverse transcription polymerase chain reaction; STING, stimulator of interferon gene.
ABT-199-Induced mtDNA release is independent of BAX/BAK
Next, we investigated how ABT-199 mediated mtDNA release to the cytosol. Previous studies have demonstrated that ABT-199 potently induces apoptosis in BCL-2-dependent hematological cancer cells. By neutralizing BCL‐2, ABT-199 triggers BAX/BAK activation, leading to mitochondrial outer membrane permeabilization and consequent cytochrome C (Cyt-C) release.25 26 Thus, we investigated whether ABT-199 could elicit apoptosis in CRC cells. The data showed that ABT-199 did not trigger apoptosis (figure 5A and B; online supplemental figure 5A), nor did it induce Cyt-C release (figure 5C). This observation is consistent with numerous previous studies showing that solid cancer cells that are less dependent on BCL-2 are usually not sensitive to ABT-199 (online supplemental figure 5B). Moreover, BAK oligomerization was not significantly affected on ABT-199 treatment (figure 5D). To directly examine the requirement of BAX/BAK in ABT-199-mediated STING signaling pathway activation, we depleted Bax and Bak in CT26 cells by CRISPR/Cas9. Additionally, we administered ABT-199 in Bak/Bax-DKO cells and fractionated cell extracts into cytosolic, mitochondrial, and nuclear fractions (figure 5E). Subsequently, we purified DNA from each fraction and quantified DNA containing specific mitochondrial genes and nuclear genes using qPCR, and the result showed that Bak/Bax knockout did not affect ABT-199-induced mtDNA release (figure 5F). We performed dsDNA reagents to detect cytosolic mtDNA levels, which was also consistent with qPCR results (online supplemental figure 5C,D). Furthermore, ABT-199-induced TBK1 and STING phosphorylation was not attenuated when Bax/Bak was knocked out by CRISPR/Cas9 (figure 5G). The BAX oligomerization inhibitor BAI1 also failed to attenuate STING phosphorylation as well (figure 5H). These data suggested that BAX/BAK was not involved in ABT-199-induced mtDNA release.

BAK/BAX was not required for ABT-199-mediated STING pathway activation. (A and B) CT26 (A) and HT29 (B) cells were treated with ABT-199 (5 µM) for 6, 12, and 24 hours, and cell apoptotic rate was measured by flow cytometry with Annexin V/PI staining. (C) CT26 cells were treated with ABT-199 (5 µM) for 24 hours, and the P-TBK1 and cytochrome C proteins expression in the Cyt and Mito fractions was detected by western blot. GAPDH and HSP60 were used as markers for Cyt and Mito fractions. (D) HT-29 cells were treated with ABT-199 (5 µM) for 24 hours, followed by the treatment of cross-linking agent EGS (250 mM, 40 min, and 37°C) to stabilize oligomers. Subsequently, the BAK oligomers were detected by western blot and quantitatively analyzed by Image J analysis. (E) Bak/Bax-DKO CT26 cells were treated with ABT-199 (5 µM) for 24 hours, then fractionated into WCE, Cyt, Mito, and Nuc fractions and subjected to analysis using western blot. GAPDH, HSP60, and H3 were used as markers for Cyt, Mito, and Nuc fractions. (F) DNA from each fraction was purified, and Dloop1, Dloop2, CytB, 16s, Hk2, Tert, Ptger2, and Ndufv1 expression levels were detected by qRT-PCR. (G) CRISPR/Cas9 control or BAK/BAX double-KO cells were treated with ABT-199 (5 µM) for 24 hours and subjected to immunoblotting for P-TBK1, TBK1, P-STING, STING, BAK, and BAX levels. The relative expression of P-TBK1 and P-STING was analyzed quantitatively by Image J analysis. (H) CT26 cells were treated with ABT-199 (5 µM) alone or together with BAI1 (2 µM) for 24 hours, and the P-TBK1, TBK1, P-STING, and STING proteins were detected by western blot. Data represented the mean±SD (A, B, and D, n=3; E, F, and G, n=4). Statistical analysis of the data was performed by Student’s t-test (two groups) and one/two-way analysis of variance with Dunnett’s post hoc test (more than two groups). n.s, no significance; qRT-PCR, quantitative reverse transcription polymerase chain reaction; STING, stimulator of interferon gene.
VDAC1 oligomerization is involved in ABT-199-mediated cGAS/STING activation
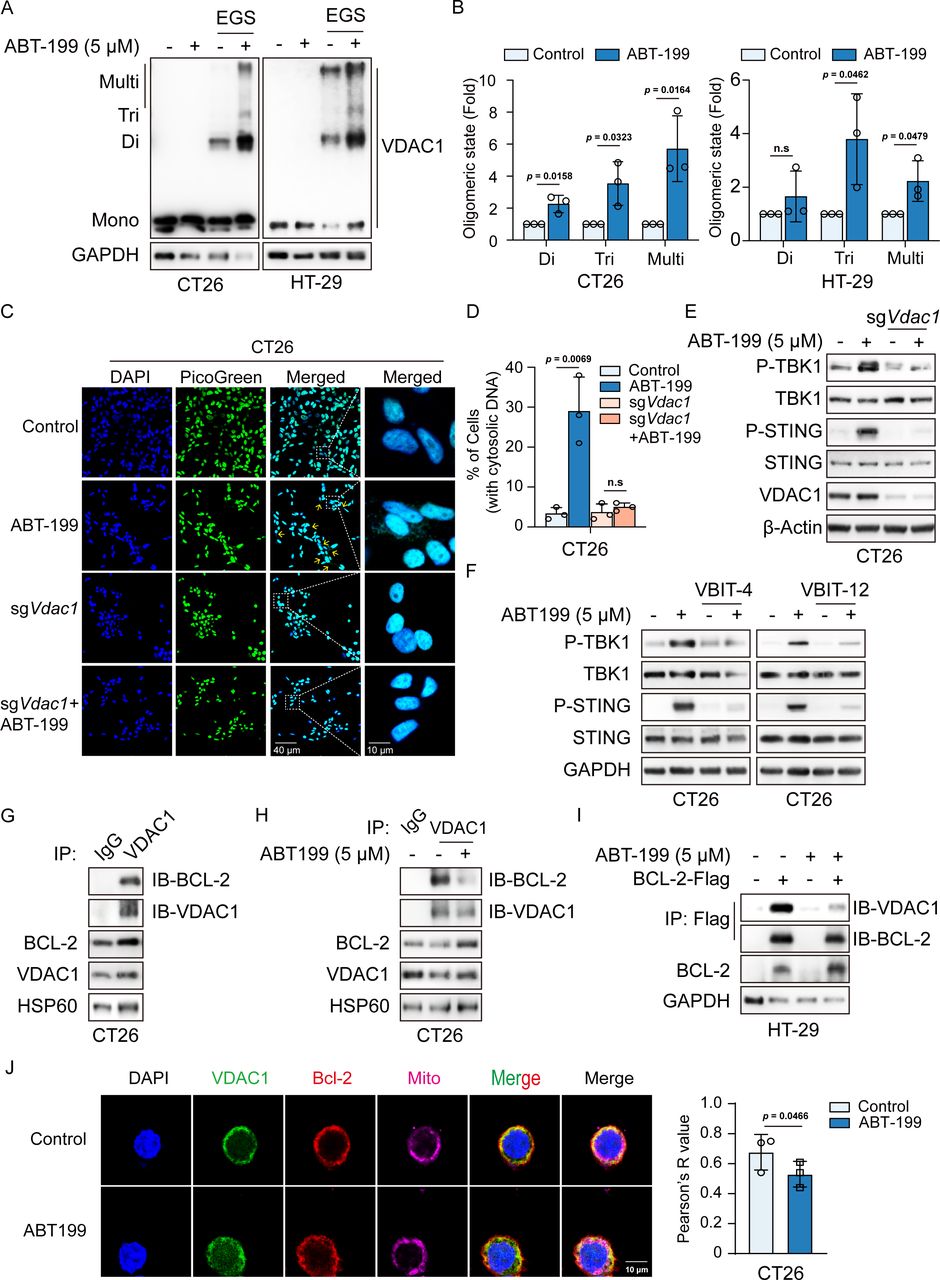
Recent studies have highlighted that mtDNA can be released by VDAC1 oligomerization to form mitochondrial pores.27 We thus assessed the role of VDAC1 in ABT-199-mediated mtDNA release. Since the oligomeric assembly of VDAC1 was required for mtDNA release, we assessed whether ABT-199 influenced the oligomeric state of VDAC1. Notably, our data revealed a significant increase in VDAC1 oligomerization on ABT-199 treatment in CT26 and HT29 cells (figure 6A and B). VDAC1 deletion abrogated ABT-199-induced increase in cytosolic dsDNA levels (figure 6C and D). In VDAC1-deficient cells, ABT-199-mediated activation of the STING signaling pathway was substantially diminished (figure 6E; online supplemental figure 6A). Furthermore, inhibition of VDAC1 oligomerization with VBIT-4 or VBIT-12 impaired ABT-199-induced TBK1 and STING phosphorylation (figure 6F). VDAC1 is a highly Ca2+-permeable channel that transports Ca2+ into the intermembrane space, thereby modulating the access of Ca2+ to transporters in the inner mitochondrial membrane.28 29 Residues E73 and E202 in VDAC1 are known to mediate calcium transport and facilitate calcium movement, and the mutation of E73 and E202 to glutamine in VDAC1 shows reduced mitochondrial Ca2+ transport.30–32 Therefore, we employed the VDAC1 E73Q/E202Q mutant to investigate whether mitochondrial calcium flow contributes to ABT-199-mediated activation of the cGAS-STING signaling pathway. VDAC1 deletion abrogated ABT-199-induced TBK1 and STING phosphorylation, while re-expression of VDAC1 WT, VDAC1 E73Q and E202Q restored ABT-199-triggered STING activation (online supplemental figure 6B). This study suggests a Ca2+-independent function of VDAC1 in ABT-induced cGAS/STING signaling.

ABT-199 induced VDAC1 oligomerization by inhibiting the interaction between BCL-2 and VDAC1. (A and B) CT26 and HT-29 cells were treated with ABT-199 (5 µM) for 24 hours, followed by the treatment of cross-linking agent EGS (250 mM, 40 min, and 37°C) to stabilize oligomers. Subsequently, the VDAC1 oligomers were detected by western blot (A) and quantitatively analyzed by Image J analysis (B). (C and D) Representative images (C) and quantitative analysis (D) of dsDNA reagent staining in CRISPR/Cas9 control or VDAC1 KO cells treated with DMSO or ABT-199 (5 µM) for 24 hours. DAPI (blue) was used to visualize the nucleus. Scale bar: 40 µm and 10 µm. (E) CRISPR/Cas9 control or VDAC1 KO cells were treated with ABT-199 (5 µM) for 24 hours and subjected to immunoblotting for P-TBK1, TBK1, P-STING, STING, and VDAC1 levels. (F) CT26 cells were treated with VBIT-4/VBIT-12 (10 µM) alone or in combination with ABT-199 (5 µM) for 24 hours, then the P-TBK1, TBK1, P-STING, and STING protein expression was measured by western blot. (G) BCL-2 bound with VDAC1 on mitochondria. Mitochondria extracted from CT26 cells were lysed and immunoprecipitated with anti-VDAC1 and anti-IgG, which was followed by immunoblotting with the BCL-2 antibody. (H) ABT-199 disrupted the endogenous interaction between BCL-2 and VDAC1. Mitochondria extracted from CT26 cells treated with ABT-199 (5 µM) for 24 hours were lysed and immunoprecipitated with anti-VDAC1 and anti-IgG, which was followed by immunoblotting with the BCL-2 antibody. (I) ABT-199 inhibited the binding of VDAC1 and BCL-2. HT-29 cells were transfected with BCL-2-Flag, and then cells were treated with ABT-199 (5 µM) for 24 hours. The cells were immunoprecipitated with anti-Flag, which was followed by immunoblotting with the VDAC1 antibody. (J) ABT-199 inhibited the binding of VDAC1 and BCL-2 at the mitochondrial membrane. CT26 cells were treated with ABT-199 (5 µM) for 24 hours, followed by staining using the MitoTracker dyes to label mitochondria, and detected the interaction between VDAC1 and BCL-2 at the mitochondrial membrane by immunofluorescence assays. Scale bar: 10 µm. Data represented the mean±SD (B, D, and J, n=3). Statistical analysis of the data was performed by Student’s t-test. DAPI, 4',6-Diamidino-2-phenylindole; DMSO, dimethyl sulfoxide; double stranded DNA, dsDNA; n.s, no significance; STING, stimulator of interferon gene; VDAC1, voltage-dependent anion channel protein 1.
Given that BCL-2 has been reported to interact with the N-terminal α-helix of VDAC1 to close the VDAC channel,33 34 we hypothesized that ABT-199 induced VDAC1 oligomerization by disrupting the interaction between BCL-2 and VDAC1. To test this possibility, we initially analyzed the binding of BCL-2 and VDAC1 on the mitochondrial membrane. The results of endogenous co-immunoprecipitation experiments confirmed that BCL-2 interacted with VDAC1 (figure 6G) and that the binding of BCL-2 to VDAC1 was disrupted by ABT-199 (figure 6H). A significant decrease in the interaction between BCL-2 and VDAC1 was observed after ABT-199 treatment, as determined by transient transfection and co-immunoprecipitation experiments (figure 6I). In addition, we performed mitochondria-specific fluorescence staining using the MitoTracker, which is used to label mitochondria, to detect the interaction between VDAC1 and BCL-2 at the mitochondrial membrane by immunofluorescence assays. Our data revealed that VDAC1 interacted with BCL-2 on the mitochondrial membrane and that ABT-199 disrupted this interaction (figure 6J). The above findings collectively demonstrated that ABT-199 induced VDAC1 oligomerization by disrupting the interaction between BCL-2 and VDAC1, leading to mtDNA release and STING signaling pathway activation.
Our previous data showed that ABT-199 treatment led to a reduction in mitochondrial membrane potential (online supplemental figure 4E). To further confirm whether ABT-199-induced mtDNA release is dependent on its mediation of mitochondrial stress, we administered mtROS generators DMNQ and mitoParaquat to CT26 cells and analyzed whether they could activate the STING pathway. The results showed that neither of them could induce the activation of the STING signaling pathway (online supplemental figure 7A), although they already caused significant mitochondrial stress as indicated by reduced mitochondrial membrane potential (online supplemental figure 7B), which suggested that mitochondrial stress is not always necessarily coupled to mtDNA release. Additionally, we further analyzed mitochondrial stress induction and STING activation in BCL-2 knockdown and VDAC1 overexpression. As shown in online supplemental figure 7C,D, knockout of BCL-2 significantly promoted the phosphorylation of STING and TBK1, without affecting the mitochondrial membrane potential level (online supplemental figure 7C). Similarly, VDAC1 overexpression did not reduce mitochondrial membrane potential but significantly induced STING signaling pathway activation (online supplemental figure 7D). Furthermore, our previous data in figure 6E and F showed that ABT-199-triggered STING signaling pathway activation was abolished when VDAC1 was knocked out, and was also attenuated in the presence of VDAC1 oligomerization inhibitors. Taken together, these findings indicated that ABT-199 triggered mtDNA release and STING signaling pathway activation mainly by disrupting the interaction between VDAC1 and BCL-2.
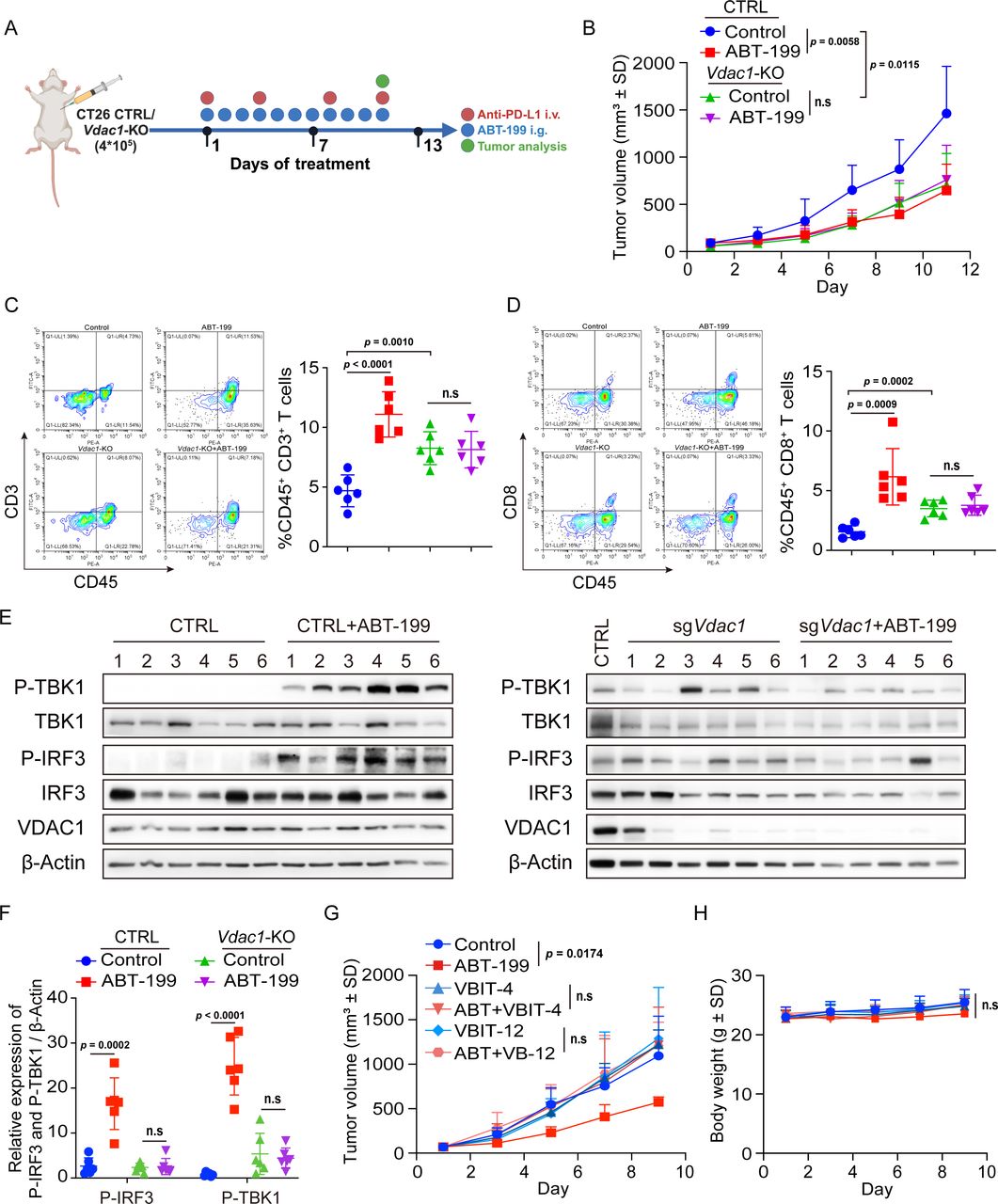
VDAC1 is essential for ABT-199-induced antitumor efficacy
To determine whether VDAC1 deficiency-induced STING pathway suppression functionally impaired ABT-199-mediated tumor growth suppression, we compared the tumor growth rates of CTRL and Vdac1-KO CT26 cells in BALB/c mice, both with and without ABT-199 treatment (figure 7A). Consistent with previous findings, ABT-199 significantly inhibited tumor growth; however, Vdac1 deficiency abrogated ABT-199-elicited antitumor immunity. Of these, the knockdown of Vdac1 similarly inhibited tumor growth compared with that in the control group (figure 7B). This observation aligns with previous reports suggesting that Vdac1 knockdown mediates tumor inhibition by reversing metabolic reprogramming.35 Subsequently, we employed flow cytometry to analyze tumor-infiltrating lymphocytes after treatment. We found a significant increase in the number of intratumoral CD3+ and CD8+ T cells, whereas this increase was abolished after Vdac1 knockdown (figure 7C and D). By analyzing STING pathway activation, we found that ABT-199-induced TBK1 and IRF3 phosphorylation was impaired by Vdac1 knockdown (figure 7E and F). In addition, we introduced a VDAC1 oligomerization inhibitor VBIT-4 to further validate the impact on the antitumor effects of ABT-199. The results showed that VBIT-4 and VBIT-12 monotherapy had no obvious effect on tumor growth, but significantly reversed the antitumor effect of ABT-199, which did not induce substantial changes in body weight (figure 7G and H). Together, these data suggested that ABT-199-triggered antitumor immunity was dependent on VDAC1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Vdac1 depletion impaired ABT-199-induced T-cell recruitment and antitumor efficacy. (A) Schematic diagram of the experimental procedure: BALB/c mice were inoculated with CTRL and Vdac1-KO CT26 cells. Then the mice were treated with ABT-199 (100 mg/kg) daily, and the tumor size, T-cell infiltration, and STING signaling pathway activation were assessed. (B) The tumor volume of BALB/c mice bearing the CTRL and Vdac1-KO CT26 cells. (C and D) The tumor-infiltrating CD45+CD3+ T cells (C) and CD45+CD8+ T cells (D) were analyzed by flow cytometry. (E and F) Tumor tissues were separated, and the expression of P-TBK1, TBK1, P-IRF3, IRF3, and VDAC1 proteins was detected by western blot. (G and H) BALB/c mice were inoculated with CT26 cells. Subsequently, the mice were treated with ABT-199 (100 mg/kg) and VBIT-4 (20 mg/kg), VBIT-12 (20 mg/kg) either individually or in combination. Tumor growth curves and body weight were assessed. Data were the mean±SD (n=6). The data were analyzed by Student’s t-test. i.g., intragastric; i.v., intravenous; n.s, no significance; PD-L1, programmed death-ligand 1; VDAC1, voltage-dependent anion channel protein 1.
Discussion
Recent studies have demonstrated that STING pathway activation is an effective immune-priming strategy to augment ICIs efficacy. Here, we demonstrated that ABT-199 triggered the activation of the STING signaling pathway in CRC cells and enhanced the immune response to ICIs by facilitating the infiltration of cytotoxic T lymphocytes. Mechanistic studies revealed that ABT-199 disrupted the interaction between BCL-2 and VDAC1, thereby restoring VDAC1 oligomerization to facilitate the release of mtDNA. Our study defines a mechanism by which BCL-2 inhibition activates the STING signaling pathway and provides a rationale for the development of the use of ABT-199 to treat solid tumors.
ABT-199, a selective and potent BCL-2 inhibitor, induces Cyt-C release and activates cysteine-containing aspartate-specific proteinase (caspase) by binding BCL-2 proteins and displacing the proapoptotic proteins BAX and BAK, and thereby initiating apoptosis. ABT-199, which is known to depend on BCL-2 for survival, has been approved for the treatment of chronic lymphocytic leukemia, small lymphocytic lymphoma, and acute myeloid leukemia; however, its efficacy in solid tumors, characterized by abundant BCL-XL and MCL-1 expression, does not hinge on BCL-2 for survival and has proven quite limited. In particular, BCL-XL plays a more important role than BCL-2 in CRC progression,36 which also explains why ABT-199 does not cause apoptosis in CRC. The data presented here suggest that ABT-199 may also have utility in additional solid tumors through its previously unappreciated ability to activate the STING signaling pathway. Approximately 80–85% of patients with CRC are considered to have “cold” tumors with MSS or low MSI that do not respond to ICIs. By activating STING signaling, ABT-199 has the potential to convert “immune cold” into “immune hot” cancer and thus could be a candidate for combination therapy with ICIs.
ABT-199 interacts with BCL-2 selectively in the BH3-binding groove to relieve the repression of BAX/BAK, which homodimerizes or heterodimerizes to permeabilize mitochondria, preventing apoptosis through the release of Cyt-C and subsequent caspase activation. mtDNA can also be released during this process; however, this release does not generally lead to STING pathway activation since activated caspases attenuate this response.21 37 Here, in CRC cells, we found that ABT-199 triggers mtDNA release to the cytoplasm when apoptosis is not induced. This mtDNA can be detected by cGAS and thus triggers the immune response. Our study also showed that ABT-199 does not activate BAX/BAK or cause Cyt-C release, further indicating that apoptosis is not induced. Findings from other studies suggest that mtDNA release could be distinct from apoptosis, as exemplified by scenarios in which IL-1β and TNF induce mitochondrial dysfunction and mtDNA release without inducing apoptosis.37 38 Thus, our findings indicate that ABT-199 could cause mtDNA release and STING activation without inducing apoptosis in solid cancer cells.
VDAC, a highly abundant mitochondrial outer membrane protein, can oligomerize to mediate the release of proapoptotic factors and mtDNA under oxidative stress conditions; among them, VDAC1 was noted for its highest abundance. Emerging studies have indicated that VDAC1 oligomerization mediates mtDNA release, thereby activating the STING pathway in conditions in which BAX/BAK is not activated. Thus, we directly examined the role of VDAC1 in the ABT-199-mediated activation of the STING pathway. Our study revealed that VDAC1 deficiency and VDAC1 oligomerization inhibition abolish the activation of the STING pathway by ABT-199 (figure 6C–F; online supplemental figure 6C,D), and that robustly represses ABT-199-mediated antitumor effects (figure 7). Our results also revealed that ABT-199 promotes VDAC1 oligomerization to release mtDNA by disrupting the interaction between BCL-2 and VDAC1 (figure 6H–J). A newly published paper reported that venetoclax (ABT-199) enhanced the function of DCs by activating the mtDNA-cGAS-STING pathway and synergized with PD-1 blockade, but the detailed mechanism remained unclear. Our study indicated that BCL2 inhibitor-triggered STING activation in DCs may induce mtDNA release by promoting VDAC1 oligomerization.
Previous reports indicate that VDAC1 oligomerization is strongly correlated with apoptosis, a process that does not activate the innate immune response. Recent research has proposed that VDAC oligomer pores mediate mtDNA release to trigger the IFN-I response under oxidative stress conditions. In addition, calcium overload initiates mPTP opening, followed by VDAC oligomerization and subsequent release of mtDNA into the cytosol, ultimately leading to STING pathway activation. Transmembrane BAX inhibitor motif containing 6 (TMBIM6) prevents VDAC1 oligomerization by binding to VDAC1, and LPS represses TMBIM6 expression and disassociates it from VDAC1, resulting in VDAC1 oligomerization.39 Additionally, ubiquitination of VDAC1 at lysine 53 by Parkin interrupts VDAC1 oligomerization and impedes mtDNA release into the cytoplasm.40 Likewise, BCL-XL has been shown to interact with VDAC1 near its dimerization interface, thereby preventing VDAC1 oligomerization,41 which is consistent with our finding that BCL-2 impedes VDAC1 oligomerization. BCL-2 interacts with the N-terminal α-helix of VDAC1 through its BH4 domain, whereas ABT-199 binds the hydrophobic BH3 binding groove of BCL-2 and prevents the binding of proapoptotic proteins.42–44 The binding of ABT-199 to the BH3 domain of BCL-2 causes a conformational change in the BH4 domain, leading to the inhibitory activity of inositol 1,4,5-trisphosphate receptor (IP3R), which binds to the BH4 domain of BCL-2.45 Therefore, we hypothesize that binding of ABT-199 to BCL-2 disrupts the interaction between BCL-2 and VDAC1 by altering the structure of BCL-2, but this hypothesis requires further experimental confirmation.
In summary, the results obtained in our study suggest that ABT-199 could induce mtDNA release to activate the STING pathway and synergize with the ICI PD-L1 antibody to achieve tumor regression. Our findings identify a small-molecule STING agonist and reveal the mechanism by which ABT-199 promotes VDAC1 oligomerization to activate the STING pathway. Taken together, our study provides a potential combined strategy for clinical immunotherapy of CRC.
Supplemental material
Supplemental material
Data availability statement
Data are available upon reasonable request. No data are available.
Ethics statements
Patient consent for publication
Ethics approval
Clinical CRC samples obtained at The Zhejiang Cancer Hospital were approved by the Ethical Committees (IRB-2019-175). Participants gave informed consent to participate in the study before taking part.
References
Footnotes
BY and LD contributed equally.
Contributors WZ: Writing—review and editing, Writing—original draft, Project administration, Investigation, Data curation. XP: Writing—original draft, Project administration, Investigation, Formal analysis. LW: Project administration, Investigation, Methodology, Data curation. WL: Investigation, Methodology, Formal analysis. XD: Software, Resources, Methodology. MZ: Methodology, Formal analysis. HG: Methodology, Formal analysis. XC: Funding acquisition, Formal analysis. YX: Resources, Methodology. HW: Software, Resources. QH: Funding acquisition, Resources, Formal analysis. BY: Writing—review and editing, Visualization, Supervision, Formal analysis, Data curation, Conceptualization. LD: Writing—review and editing, Funding acquisition, Supervision, Formal analysis, Data curation, Conceptualization. LD acts as the guarantor of the study.
Funding This work was supported by the National Natural Science Foundation of China (No.82273949 to LD, No.82330114 to QH) and the Zhejiang Provincial Natural Science Foundation of China (No. LD25H300002 to LD).
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.