Article Text

Abstract
Background SOT201 and its murine surrogate mSOT201 are novel cis-acting immunocytokines consisting of a humanized/murinized/, Fc-silenced anti-programmed cell death protein 1 (PD-1) monoclonal antibody (mAb) fused to an attenuated human interleukin (IL)-15 and the IL-15Rα sushi+ domain. Murine mPD1-IL2v is a conjugate of a murinized, Fc silenced anti-PD-1 mAb bearing human IL-2 with abolished IL-2Rα binding. These immunocytokines spatiotemporally reinvigorate PD-1+ CD8+ tumor-infiltrating lymphocytes (TILs) via cis-activation and concomitantly activate the innate immunity via IL-2/15Rβγ signaling.
Methods Human peripheral blood mononuclear cell and cell lines were used to evaluate cis/trans activity of SOT201. Anti-PD-1 mAb responsive (MC38, CT26) and resistant (B16F10, CT26 STK11 KO) mouse tumor models were used to determine the anticancer efficacy, and the underlying immune cell activity was analyzed via single-cell RNA sequencing and flow cytometry. The expansion of tumor antigen-specific CD8+ T cells by mSOT201 or mPD1-IL2v and memory CD8+ T-cell generation in vivo was determined by flow cytometry.
Results SOT201 delivers attenuated IL-15 to PD-1+ T cells via cis-presentation, reinvigorates exhausted human T cells and induces higher interferon-γ production than pembrolizumab in vitro. mSOT201 administered as a single dose exhibits strong antitumor efficacy with several complete responses in all tested mouse tumor models. While mPD1-IL2v activates CD8+ T cells with a 50-fold higher potency than mSOT201 in vitro, mSOT201 more effectively reactivates effector exhausted CD8+ T cells (Tex), which demonstrate higher cytotoxicity, lower exhaustion and lower immune checkpoint transcriptional signatures in comparison to mPD1-IL2v in MC38 tumors in vivo. This can be correlated with a higher rate of complete responses in the MC38 tumor model following mSOT201 treatment when compared with mPD1-IL2v. mSOT201 increased the relative number of tumor antigen-specific CD8+ T cells, and unlike mPD1-IL2v stimulated greater expansion of adoptively transferred ovalbumin-primed CD8+ T cells simultaneously limiting the peripheral CD8+ T-cell sink, leading to the development of memory CD8+ T cells in vivo.
Conclusions SOT201 represents a promising therapeutic candidate that preferentially targets PD-1+ TILs, delivering balanced cytokine activity for reviving CD8+ Tex cells in tumors. SOT201 is currently being evaluated in the Phase I clinical study VICTORIA-01 (NCT06163391) in patients with advanced metastatic cancer.
- Cytokine
- Immune Checkpoint Inhibitor
- Immunotherapy
- T cell
- Tumor microenvironment - TME
Data availability statement
Data are available upon reasonable request. Data are proprietary of SOTIO Biotech AG and available on reasonable request. The datasets generated and/or analysed during the current study are available from the corresponding authors on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The cis-acting anti-programmed cell death protein 1 (PD-1) antibody-based immunocytokines bearing interleukin (IL)-2 or IL-15 represent promising immunotherapeutics currently being tested in clinical trials. These compounds have the potential to reinvigorate exhausted CD8+ T cells in the tumor microenvironment.
WHAT THIS STUDY ADDS
Here, we newly highlighted that the strength of IL-2/15Rβγ engagement by cis-targeting PD-1-IL-2/15 immunocytokines is the key determinant of antitumor efficacy. SOT201, a novel cis-targeting anti-PD-1 antibody-based IL-15 immunocytokine with attenuated activity enhances the cytotoxicity of effector exhausted CD8+ T cells in tumors leading to superior antitumor efficacy compared with mPD1-IL2v, an IL-2/15Rβγ agonist that is over 50-fold more potent.
HOW THIS STUDY MIGHT AFFECT RESEARCH PRACTICE OR POLICY
Understanding how attenuated immunocytokines can improve the quality and revert the phenotype of exhausted tumor antigen-specific CD8+ T cells via modulation of their affinity to IL-2/15Rβγ and cytokine signaling strength represents an important step towards improving the final therapeutic effect.
Introduction
Interleukin-15 (IL-15) and interleukin-2 (IL-2) are crucial for activation and cytotoxicity of natural killer (NK) and CD8+ T cells; however, these cytokines are not functionally redundant.1–3 IL-2 favors regulatory T cell (Treg) homeostasis whereas IL-15 predominantly expands memory CD8+ T cells, NK cells, and natural killer T (NKT) cells. Both IL-15 and IL-2 signal through a receptor sharing the IL-2/15 receptor β (IL-2/15Rβ, CD122) and the common gamma chain (γc or γ, CD132), activating the Jak-STAT, PI3K-Akt, and Ras-MAPK pathways. However, both cytokines have distinct alpha-receptor chains, IL-2Rα (CD25) for IL-2 and IL-15Rα (CD215) for IL-15, that determine cellular responsiveness. IL-15 is dominantly presented by antigen-presenting cells in trans bound to IL-15Rα to the IL-2/15Rβγ on the target cells, whereas IL-2 is presented in cis to IL-2Rαβγ on the same cell surface.1 3 4 IL-15-based and IL-2-based therapeutics showed limited success in clinical trials.5–8 This may be attributed to limited tumor delivery at doses inducing systemic dose-limiting toxicities which result mainly from NK and CD8+ T-cell activation in the periphery.8 9 The therapeutic index of cytokines may be improved by their targeting to the tumor microenvironment via tumor-antigen specific antibodies and/or by decreasing their affinity to the target receptors through cytokine mutation or masking of the cytokine binding site.8 9
Specifically, IL-15 and IL-2-based conjugates of antibodies against immune checkpoints such as programmed cell death 1 (PD-1), which is highly expressed on CD8+ tumor-infiltrating lymphocytes (TILs), displayed prominent antitumor efficacy in murine tumor models thereby showing a potential to extend the rate of objective responses over checkpoint inhibitor standalone immunotherapy.10–15 Immune checkpoint inhibitors (CPIs) targeting PD-1 or its ligand (PD-L1) are now a standard of care for many advanced cancers.16 However, most patients respond transiently and do not display long-term benefits due to primary or secondary resistance.17 Notably, the low infiltration and the exhaustion of CD8+ TILs represent the major hurdle in stimulating effective antitumor immunity.18 Exhausted CD8+ T cells can be divided into precursor exhausted T cells (Tpex) and terminally exhausted T cells (Tex), both consisting of functional subtypes with a varying capacity to acquire T-cell effector functions.19–23 These exhausted T cells, which can regain high cytotoxic activity in tumors were designated as better effector CD8+ T cells.10 24 Whereas PD-1/PD-L1 blockade was shown to expand and differentiate better effector Tpex, they did not activate Tex subsets.21–23 25 Several anti-PD-1-IL-2/15-based immunocytokines were shown to reinvigorate both exhausted CD8+ T-cell populations Tpex and Tex in preclinical models and human tumor tissues ex vivo,10–12 14 however these studies only compared respective anti-PD-1 monoclonal antibodies (mAb), IL-2/15 cytokine monotherapy and their combination.10–12 14
This study is the first to directly compare IL-2 and IL-15-based anti-PD-1 immunocytokines, specifically correlating the quality of reinvigorated exhausted CD8+ T cells to the immunocytokine-mediated antitumor efficacy. We characterized SOT201, a novel immunocytokine with optimized potency composed of an anti-PD-1 mAb and activity-attenuated human IL-15 fused to the IL-15Rα sushi+ domain (RLI-15m). We demonstrate SOT201 cis-targeting of PD-1+CD8+ T cells by engaging both the IL-2/15Rβγ and PD-1 to provide downstream signaling in vitro and in vivo. Importantly, SOT201 was compared with another clinical stage PD1-IL2v immunocytokine which also signals via IL-2/15Rβγ.26 Murine surrogate mPD1-IL2v consisting of an anti-PD-1 mAb genetically fused to IL-2 cytokine with abolished IL-2Rα binding, showed 50-fold higher activity on CD8+ T cells compared with the murine surrogate of SOT201, mSOT201. In the MC38 mouse tumor model, we showed that mSOT201 induces superior reinvigoration of the CD8+ Tex cells with higher cytotoxicity/proliferation and lower exhaustion/immune checkpoint transcriptional signatures in tumors compared with mPD1-IL2v. This correlated with more durable antitumor efficacy. This suggests that PD-1-targeted delivery of an activity-attenuated IL-15 to IL-2/15Rβγ limits the peripheral sink leading to a longer half-life, while polarizing CD8+ T cells in tumors towards a higher effector quality via cis-activation.
Materials and methods
Immunocytokines, cytokines, antibodies and human cancer cell lines
The molecular characteristics of tested cytokines, immunocytokines and antibodies are described in online supplemental table 1 and detailed in international patent application WO2022/268983A2. Kit225 wt27 was cultivated in Roswell Park Memorial Institute 1640 medium (RPMI 1640) with 6% heat-inactivated fetal bovine serum (FBS) (Sigma Aldrich, USA), 1% GlutaMAX, 1% PenStrep and 5 ng/mL of IL-2. Kit225-PD-1+ cell line was prepared using plasmid encoding human PD-1 (VectorBuilder, Germany) selected with 1 µg/mL of puromycin. Raji wt obtained from the American Type Culture Collection, USA, and Raji PD-1+ cells from InvivoGen, USA, were cultivated in Iscove’s Modified Dulbecco’s Medium (IMDM), 10% FBS, 1% PenStrep and 1% normocin (InvivoGen, USA). Raji-PD-1+ cell medium was supplemented with 2 µg/mL blasticidin (InvivoGen, USA). All cell lines were cultivated at 37°C, 5% CO2. Reagents were from Gibco, Thermo Fisher Scientific, USA, unless stated otherwise.
Supplemental material
Cell proliferation and STAT5 phosphorylation assay
Kit225 wt or Kit225 PD-1+ cells were treated with SOT201 for 72 hours at 37°C with the last 6 hours addition of 10% Alamar Blue (Bio-Rad, USA) to detect proliferation at A570 nm by Spark reader (TECAN). For STAT5 phosphorylation, cells stained with a CellTrace Violet Cell Proliferation Kit (Invitrogen, USA) were incubated with SOT201 at the indicated concentrations for 15 min at 37°C followed by anti-pSTAT5-Alexa Fluor 647 antibody (BD Biosciences, USA). Pembrolizumab (Keytruda, Merck) was incubated for 30 min at 37°C. For the trans activation, Raji wt or Raji PD-1+ cells were coincubated with SOT201 for 30 min before the addition of Kit225 wt for 15 min. Data were collected using the flow cytometer LSRFortessa (BD Biosciences, USA) and evaluated using BD Diva (BD Biosciences, USA) and FlowJo (Tree Star, USA).
Exhausted T-cell restimulation assay
T cells were isolated from human peripheral blood mononuclear cell (hPBMC) using a Pan T Cell Isolation Kit (Miltenyi Biotec, Germany). For exhaustion, T cells were cultivated in RPMI 1640 with 10% FBS, 1 mM sodium pyruvate, 1% non-essential amino acids (NEAA), 50 mM 2-mercaptoethanol, 2 mM L-glutamine and 1% gentamycin (all Thermo Fisher Scientific, USA) together with 25 µL ImmunoCult Human CD3/CD28 T Cell Activator (STEMCELL Technologies, Canada) for 7 days with a fresh restimulation on day 2 and 4. The T-cell phenotype was confirmed by flow cytometry (online supplemental table 2). For the T-cell restimulation assay, T cells were mixed with autologous dendritic cells (DCs) (10:1) and treated with SOT201 at 100 or 10 nM, pembrolizumab or IgG4 isotype (BD, USA) at 10 µg/mL (60 nM) and the Staphylococcal enterotoxin B (Sigma Aldrich, USA) at 0.0001 µg/mL at 37°C. After 72 hours, the production of interferon (IFN)-γ, tumor necrosis factor (TNF)-α and granzyme B in cell culture supernatants was analyzed using a Luminex kit (R&D Systems, USA) by flow cytometry (LSRFortessa).
Human PBMC potency assay
hPBMCs (four to eight donors) were isolated using Ficoll-Paque gradient centrifugation (Thermo Fisher Scientific, USA). hPBMCs were cultivated in RPMI 1640 with 2 mM GlutaMAX I CTS, 1% PenStrep, 1 mM sodium pyruvate, 1% NEAA, 50 mM 2-mercaptoethanol (all Thermo Fisher Scientific, USA) and 10% human AB serum (heat-inactivated, Invitrogen, USA) and incubated with SOT201 at 0.0001–100 nM for 7 days, 37°C. The immune cell proliferation (Ki67+) and phenotype (online supplemental table 2) were assessed by flow cytometry (Cytek Aurora). For PD-1 detection pembrolizumab followed by anti-human IgG-PE was used (BioLegend, USA).
PD-1/PD-L1 blocking assay and mixed lymphocyte reaction
SOT201 and pembrolizumab blocking capacity was evaluated by PD-1/PD-L1 Blockade Bioassay (Promega, USA) according to the manufacturer’s instructions. For mixed lymphocyte reaction (MLR), hPBMC from two different donors (12 pairs) were mixed in a ratio of 1:1 and co-cultured with 1 nm of SOT201 and pembrolizumab. The IFN-γ production was detected in cell culture supernatants by an IFN-γ ELISA kit (Invitrogen, USA) after 6 days.
Pharmacodynamic activity in vitro and in vivo
Spleens (BALB/c mice) were dissociated using the gentleMACS Dissociator (Miltenyi Biotec, Germany). Splenocytes were cultivated in RPMI 1640 with 10% FBS, 1% PenStrep and 1% GlutaMAX I CTS (all Thermo Fisher Scientific, USA). mSOT201 or mPD1-IL2v at 0.001 nM to 10 µM was added for 5 days, 37°C. For in vivo, C57BL/6 mice were injected intravenously with mSOT201 hPD1-mSOT201 or intraperitoneal (i.p) mPD1 (5 mg/kg), or mPD1-IL2v (0.25 mg/kg) in 0.9% NaCl (Sigma Aldrich, USA) on day 1 (two animals/group). NK and CD8+ T-cell proliferation (Ki67+) (online supplemental table 3) was detected by BD FACSymphony A5 on day 5.
Antitumor efficacy in vivo
Naïve C57BL/6, C57BL/6-Pdcd1 tm1(hPDCD1)Smoc mice and Balb/c female mice 8–12 weeks (8–14/group) were obtained from Shanghai Lingchang Biotechnology (Shanghai, China) or breeding core at the Institute of Microbiology or Molecular Genetics of the Czech Academy of Sciences. Mice were accommodated max. n=5 per cage and acclimatized for at least 2 weeks. Mice were inoculated subcutaneously in the rear flank with MC38, MC38 hPD-L1 (1×106), B16-F10, CT26-STK11 KO (2×105) or CT26 (5×105) cells in 0.1 mL of phosphate-buffered saline. Once tumors reached 50–150 mm³, mice were randomized based on body weight to ensure group comparability using a computer-generated sequence. Mice were treated intravenously on day 0 with the compounds SOT201 (10 mg/kg), mSOT201 (5, 10 or 20 mg/kg), hPD1-mSOT201 (5 or 10 mg/kg) or mPD1-IL2v (0.25, 0.5, 1 or 2 mg/kg) in 0.9% NaCl (Sigma Aldrich, USA). The anti-mPD1-mIgG1e3 InvivoFit (mPD1) (Invivogen, France) was administered i.p on day 0, 3, 6, 9 or intravenously on day 0 at 5 or 10 mg/kg, or equimolarly to mSOT201 for RLI-15m (intravenous, 0.64 mg/kg). The antitumor efficacy was assessed in comparison to control untreated tumor-bearing animals. The MC38 rechallenge was done using naïve or cured mice after 100 days. For depletion, anti-CD8 antibody (clone: 2.43, Bio X Cell, USA) was administered on days −4 to –1, +1, +5 i.p, 100 µg/mouse. The immune cells in tumors and spleen were analyzed by immunophenotyping (online supplemental table 3) using BD FACSymphony A5. In all studies, humane endpoints were predefined in accordance with ethical guidelines. Mice were euthanized if tumors exceeded 3,000 mm³, ulceration or skin penetration occurred, or body weight loss surpassed 20%. In addition, clinical condition and mice weight were monitored daily or every other day, respectively. At termination, animals were euthanized via CO₂ inhalation, and tumors were measured twice weekly using calipers with volume calculated as V=0.5×a×b². All studies adhere to the ARRIVE guidelines for animal research reporting.
Pharmacokinetic activity in vivo
Serum from two naïve C57BL/6 mice/time point was collected at 0, 15 min, 2 hour, 8 hours, 24 hours, 48 hours, 96 hours and 192 hours after the administration, and analyzed by ELISA using anti-SOT201 rabbit polyclonal antibody (Agro-Bio, France) and anti-human IL-15 biotinylated antibody (BAM247, R&D Systems, USA), mouse PD-1 biotinylated protein (50124-M08H-B) and rabbit polyclonal anti-IL-2 HRP antibody (11848-RP01-H) (both SinoBiological, China). The parameters were assessed by Phoenix WinNonlin (Certara, USA).
Antigen-specific CD8+ T cells and memory T cells
For tumor antigen-specific T cells, CD45+ TILs were isolated from MC38 tumors 5 days after treatments using CD45 MicroBeads (Miltenyi Biotec, Germany). Cells were stained with a 10 µL RPL18 Dextramer (Immudex, Denmark), Live/Dead dye and other antibodies (online supplemental table 3). For adoptive T-cell transfer, 3×105 isolated OT-I CD8+ T cells from spleens of OT-I/Rag1−/−/Ly5.1+ mice were intravenously injected into C57BL/6 (Ly5.2+) mice on day 0. On day 1, mice were injected i.p with 350 µg/mouse of ovalbumin (OVA; Worthington, USA). After 24 hours, mSOT201, hPD1-mSOT201 (both 5 mg/kg, intravenously) or mPD1-IL2v (0.5 mg/kg intravenous) were administered to OVA-injected or non-injected mice (3–5 animals/group). Spleens harvested on days 5 and 47 were analyzed by flow cytometry (online supplemental table 3). For memory T-cell phenotype and functionality, splenocytes isolated on day 47 were incubated with 10 µM SIINFEKL (GenScript, USA) for 6 hours and with Brefeldin A (1 µg/mL, eBioscience, USA) the last 4.5 hours. The intracellular TNF-α and IFN-γ as well as CD44 and CD122 were analyzed by flow cytometry (BD FACSymphony A5).
Single-cell RNA sequencing (scRNA-seq) analyses
scRNA-seq was performed from freshly isolated CD45+ MC38 TILs using the Chromium Next GEM Single Cell 3’ Reagent Kits V.3.1 kit and the Illumina NovaSeq X Plus platform. scRNA data were processed with the Cell Ranger pipeline (V.7.2.0) and via the Seurat (V.4.3).28 Briefly, features found in a minimum of five cells were kept, cells with at least 200 unique features detected were kept. Cells enriched with ribosomal/mitochondrial genes were also excluded from the analysis. Multiplets were filtered out via DoubletFinder (V.2.0.3).29 Unsupervised clustering was performed using a graph-based clustering approach with the Louvain algorithm. Uniform Manifold Approximation and Projection was then applied. For cell type annotation the CellID (V.1.21),30 supplemented by manual annotation, was applied. This process involved identifying differentially expressed genes among clusters and selecting markers from PanglaoDB.31 T-cell subtypes were predicted via ProjecTILs (V.3.0.0).24 UCell package (V.2.6.2)32 was used for single-cell gene signature scores. Memory/better-effector, stem-like, naïve-like and exhausted subpopulations were identified in single cell clusters with profound CD3g expression (excluding Treg) based on markers10 (online supplemental table 4).
Statistical analysis
Wilcoxon signed-rank test, unpaired t-test or one-way analysis of variance were applied using GraphPad V.10 (San Diego, California, USA) and R (https://www.r-project.org/) software. Statistical significance ****p<0.0001, ***p<0.001 **p<0.01, *p<0.05.
Results
SOT201 is a cis-acting, PD-1-targeted and activity-optimized IL-15 immunocytokine that blocks PD-1/PD-L1, enhances IFN-γ production and reinvigorates exhausted human T cells in vitro
SOT201 consists of a humanized anti-PD-1, Fc-silenced (L235E)33 IgG4 mAb genetically fused to a single molecule of RLI-15 bearing a N65A substitution (RLI-15m) (figure 1A, online supplemental table 1). SOT201 also contains an S228P substitution preventing IgG4 Fab-arm exchange.34 RLI-15 is a fusion of a human IL-15 linked with a 20 amino acid linker to the C-terminus of the IL-15Rα sushi+ domain.35–38 The N65A substitution in the IL-15 sequence was selected to attenuate binding to the IL-2/15Rβγ and results in lower cytokine activity on target cells (figure 1B). SOT201-induced proliferation of IL-2/15Rαβγ+ Kit225 cells (Kit225 wt) (figure 1B) was 36-fold lower compared with cells stimulated with SOT201 wt, an SOT201 surrogate molecule containing RLI-15 without the N65A substitution in IL-15, and 323-fold lower compared with SOT101, the wildtype RLI-15 molecule (online supplemental table 1)35–37 (figure 1B). Of note, a ninefold decrease in SOT201 wt-induced proliferation of Kit225 in comparison to SOT101 was observed, suggesting that the attachment of RLI-15 to the C-terminus of the anti-PD-1 mAb decreases the cytokine’s potency.

Cis-acting SOT201 blocks PD-1/PD-L1 interactions, enhances IFN-γ production and reinvigorates partially exhausted human T cells in vitro. (A) Schematic representation of SOT201 molecule consisting of a humanized, Fc-silenced (L235E) IgG4 monoclonal antibody against PD-1 fused to one molecule of RLI-15 bearing N65A mutation. (B) Kit225 wt (expressing IL-2/15Rαβγ only) proliferation induced by SOT201, SOT201 wt (SOT201 without N65A mutation) and SOT101 (naked RLI-15, a complex of a human IL-15 linked to the IL-15Rα sushi+ domain). (C) Cis-acting SOT201 induces proliferation of Kit225 expressing PD-1+ and IL-2/15Rαβγ (Kit225 PD-1+). (D) Pembrolizumab blocks the SOT201-induced STAT-5 phosphorylation in Kit225-PD-1+. (E) No SOT201-induced STAT-5 phosphorylation in Kit225 wt in trans when co-incubated with Raji PD-1+ and lacking IL-2/15Rβγ. Representative of n=2–3. Schemes were created with BioRender.com. (F) SOT201-induced proliferation of PD-1+ and PD-1− hPBMC immune cell populations. Mean±SEM from 4 to 8 donors. (G) SOT201 blocks PD-1/PD-L1 interactions in vitro with IC50 of 1 nM (PD-L1 aAPC/CHO-K1 reporter cells). (H) SOT201 enhances IFN-γ production at 1 nM in mixed lymphocyte reaction (hPBMC) after 5 days in vitro. Mean±SEM of 12 donor pairs. (I) SOT201 reinvigorates partially exhausted human T cells in vitro. Means±SEM of 3 donors. hPBMC, human peripheral blood mononuclear cell; IFN, interferon; IL, interleukin; NK, natural killer; PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; SEB, Staphylococcus aureus enterotoxin B; TIL, tumor-infiltrating lymphocyte; Treg, regulatory T cell.
However, when SOT201 was tested on PD-1+ IL-2/15Rαβγ+ Kit225 cells (Kit225 PD-1+), it induced cell proliferation with a 2400-fold higher sensitivity compared with Kit225 wt (figure 1C). This response is attributed to cis-binding to PD-1 and IL-15 signaling via IL-2/15Rβγ on a single cell. The highly abundant PD-1 molecule on the surface of Kit225 PD-1+ cells is assumed to serve as a docking site for SOT201 leading to IL-2/15Rβγ signaling. This was further confirmed by testing RLI-15m, an attenuated RLI-15 bearing the N65A substitution (online supplemental table 1), which induced a comparable proliferation of both cell lines independent of PD-1 expression (figure 1C), and pembrolizumab which inhibited the SOT201-induced STAT5 phosphorylation in Kit225 PD-1+ cells (figure 1D). Trans-activation was ruled out as a major cell activation mechanism as similar levels of SOT201-induced STAT-5 phosphorylation were observed in Kit225 wt cells when co-incubated with Raji PD-1+ or Raji wt cells (figure 1E). hPBMCs were used to corroborate our findings on cells with physiological PD-1 expression (online supplemental figure 1A). SOT201 induced a significantly higher proliferation of CD8+, CD4+, memory CD8+, and CD4+ T cells, but not NK cells and Tregs in PD-1+ in comparison to PD-1− subpopulations, specifically at low concentrations (figure 1F, EC50 values in online supplemental table 5). At high SOT201 concentrations, the stimulatory effect became less dependent on PD-1, as the non-PD-1-targeted attenuated RLI-15 bearing isotype control (anti-digoxigenin human IgG1 bearing LALAPG substitutions39 online supplemental table 1, figure 1F) showed comparable activation of target cells only at high concentrations. In functional assays, SOT201 blocked the PD-1/PD-L1 interaction at an IC50 of 1 nM which was comparable to pembrolizumab (figure 1G) and induced a significantly higher IFN-γ production over pembrolizumab in an MLR assay (figure 1H). SOT201 also stimulated the production of TNF-α, IFN-γ, and granzyme B from partially exhausted human T cells in vitro (figure 1I) (online supplemental figure 1B,C). Collectively, this data demonstrates that SOT201 delivers attenuated IL-15 via cis to PD-1+ T cells favoring CD8+ and CD4+ T cells over NK cells and Tregs. This results in enhanced IFN-γ production and reinvigoration of partially exhausted human T cells in vitro.
SOT201 and its mouse surrogate mSOT201 induce strong antitumor efficacy in anti-PD-1 mAb sensitive and resistant mouse tumor models via cis activation in vivo
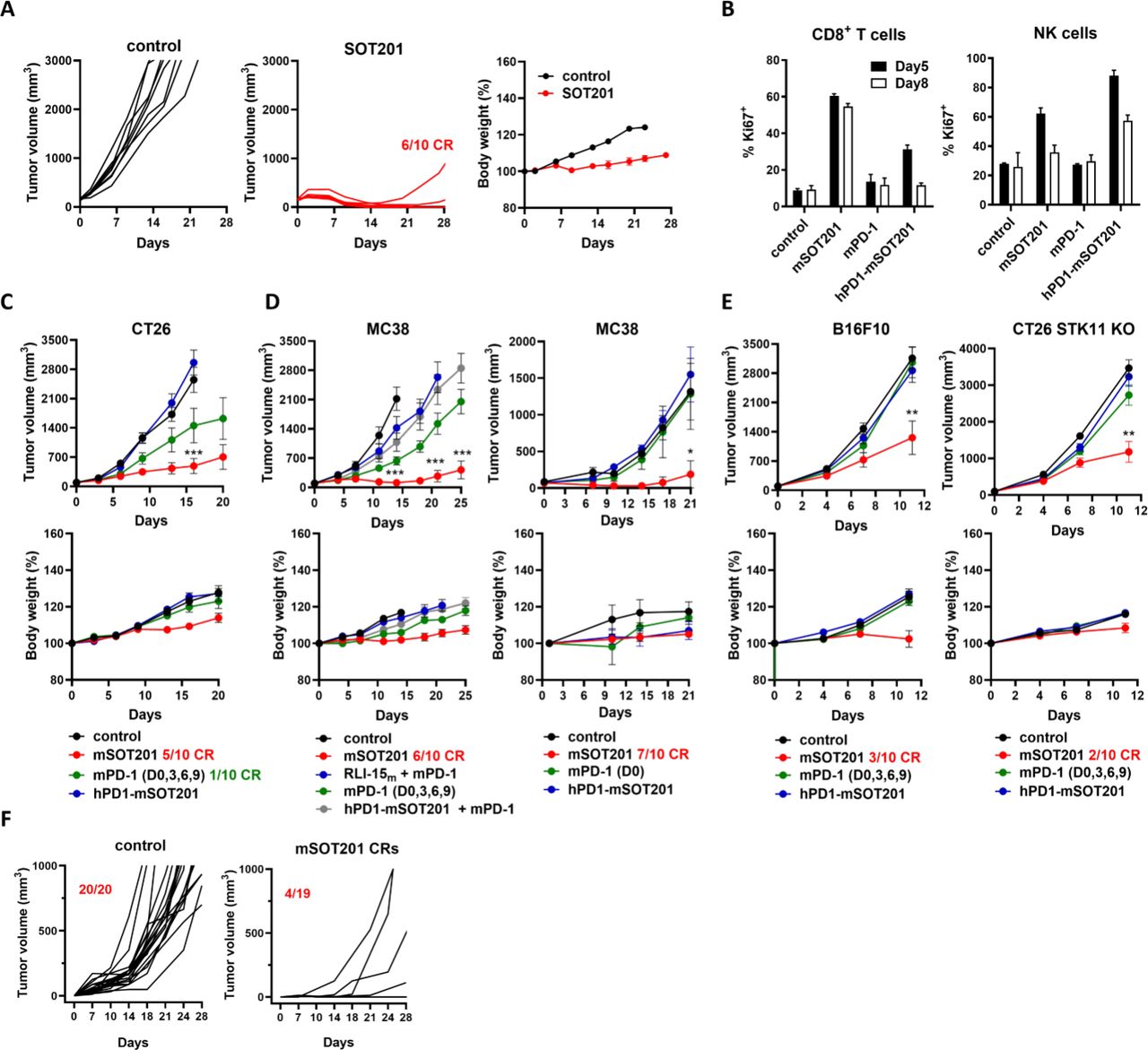
SOT201 demonstrated strong antitumor efficacy in hPD1 transgenic mice bearing MC38-hPD-L1 tumors without inducing body weight loss (figure 2A). To confirm the cis-activation by SOT201 in vivo, mouse surrogates were generated: mSOT201 consisting of anti-mouse PD-1 mouse IgG1 mAb clone RMP1-14 bearing the D265A silencing substitution in the Fc region40 genetically fused with a single molecule of attenuated RLI-15m, and hPD1-mSOT201 consisting of anti-human PD-1 mouse IgG1 clone RMP1-14 bearing the D265A mutation in the Fc region with the attenuated RLI-15m (online supplemental table 1). hPD1-mSOT201, which targets human PD-1 and is not cross-reactive to murine PD-1 served as non-PD-1 binding attenuated cytokine activity control (online supplemental table 1). mSOT201 elicited strong CD8+ T cell and weaker NK cell proliferation at days 5 and 8 in contrast to hPD1-mSOT201, which resulted in weak CD8+ T cell and strong NK cell proliferation, whereas the mPD-1 (anti-mouse PD-1 RMP1-14 clone mAb) did not induce cell proliferation (figure 2B). Importantly, mSOT201 showed superior antitumor efficacy in CT26 (figure 2C) and MC38 mouse models (figure 2D) in comparison to hPD1-mSOT201 and mPD-1 when administered at equimolar dose. mPD-1 dosed four times on days 0, 3, 6, 9 improved antitumor efficacy in the MC38 model; however, only mSOT201 induced a strong antitumor effect with a high number of complete responses. The antitumor efficacy was also not improved when an equimolar dose of attenuated RLI-15m was combined with mPD-1, or when the hPD1-mSOT201 and mPD-1 combination was administered, further confirming cis-activation of target cells by SOT201 (figure 2D). Further, mSOT201 displayed superior activity compared with all compounds tested in anti-PD-1 resistant models (figure 2E). At the same time, no significant body weight loss was observed at selected doses in all models. mSOT201 elicited long-lasting immunity as 80% of mice cured of MC38 tumors were immune to a tumor re-challenge (figure 2F). Altogether, a single dose of mSOT201 can achieve highly curative effects in anti-PD-1 sensitive models via cis-activation with up to 70% of complete responses and up to 30% of complete responses in anti-PD-1 resistant mouse tumor models.

SOT201 and mSOT201 induce strong antitumor efficacy in anti-PD-1 sensitive and resistant mouse tumor models via cis activation in vivo. (A) SOT201 (10 mg/kg, intravenous) or control treatment in hPD-1 transgenic mice bearing MC38-hPD-L1 tumors. (B) Splenic CD8+ and NK cell proliferation (Ki67+) in vivo 5 days after mSOT201, hPD1-mSOT201 (anti-human PD-1 mouse IgG1 clone RMP1-14 bearing D265A mutation in Fc region with attenuated RLI-15) and mPD-1 (clone RMP1-14) treatment at 5 mg/kg intravenously. Representative of n=2, two animals/group. SOT201 efficacy in anti-PD-1 sensitive mouse models. (C) CT26 (10 mg/kg all compounds), (D) MC38 (5 mg/kg mSOT201 or equimolar to the other compounds) and (E) in anti-PD-1 resistant mouse tumor models B16F10 or CT26 STK11 KO (10 mg/kg all compounds). Representative experiment, n=8–10 animals/group. (F) Rechallenge of naïve and SOT201-cured mice (after 100 days) with MC38 tumor cells (pool of experiments n=3). The treatment is day 0 if not stated otherwise. NK, natural killer; PD-1, programmed cell death 1.
mSOT201 expands cytotoxic CD8+ Tex with a better effector phenotype and γδ T cells in MC38 tumors
To investigate the differences in antitumor efficacy, scRNA-seq from intratumoral CD45+ cells isolated 5 days after mSOT201, hPD1-mSOT201 and mPD-1 treatment were performed (figure 3A–D). ProjecTILs was used for cell identification and clustering24 (figure 3A and B, online supplemental figure 2, table 3). All compounds predominantly increased CD8+ Tex cells: mSOT201 (55.3%), mPD-1 (44.6%) and hPD1-mSOT201 (32%) compared with non-treated tumors (14.8%) (figure 3B). Unlike mSOT201, mPD-1 also expanded Tpex and CD8+ effector memory cells whereas hPD1-mSOT201 expanded predominantly CD8+ naïve-like and CD8+ early activated populations (figure 3B). Interestingly, mSOT201 expanded γδ T cells (18.2%), which was not seen with any other agent tested (≤3.6%). CD4+ T cells and Tregs were proportionally decreased in all treated samples, mainly in those treated with mSOT201 (figure 3B). Importantly, mSOT201 significantly increased the expression of cytotoxicity genes while concomitantly decreasing genes encoding immune checkpoints and exhaustion transcription factors in comparison to other treatment groups (figure 3C and D). Flow cytometry phenotyping in tumors showed increased numbers of CD8+ and γδ T cells, but not NK cells on mSOT201 treatment compared with the control and other treatments (figure 3E). In the spleen, a significantly higher increase in γδ T cell numbers was observed with mSOT201 compared with hPD1-mSOT201 suggesting that a cis-activation mechanism may also direct γδ T cell expansion. NK cells were expanded preferentially in tumors and spleens treated with hPD1-mSOT201. A slight increase in peripheral Treg numbers was only detected on mSOT201 treatment (figure 3E). In summary, the emergence of better effector CD8+ Tex cells in MC38 tumors after mSOT201-mediated cis-activation is characterized by higher cytotoxicity and lower exhaustion profiles driving superior antitumor efficacy compared with mPD-1 and other controls. Uniquely, mSOT201 expanded γδ T cells in tumors and spleen, a response not observed after treatment with any other agents tested.

mSOT201 expands cytotoxic CD8+ Tex with a better effector phenotype and γδ T cells in MC38 tumors. (A) UMAP plot of T-cell populations and γδ T cells in MC38 tumors 5 days after treatment with mSOT201, mPD-1 or hPD1-mSOT201 (5 mg/kg, intravenously). (B) Proportion of distinct T cell populations and γδ T cells. (C) Violin plots of selected genes in CD8+ Tex. (D) Violin gene signatures in CD8+ Tex. (E) The immune cells in tumors and spleen of MC38 tumor-bearing mice at day 5 post-treatment determined by flow cytometry. Mean±SEM. NK, natural killer; PD-1, programmed cell death 1; Tex, exhausted T cells; Treg, regulatory T cell; UMAP, Uniform Manifold Approximation and Projection.
mSOT201 induces better effector CD8+ Tex driving a superior antitumor response compared with mPD1-IL2v
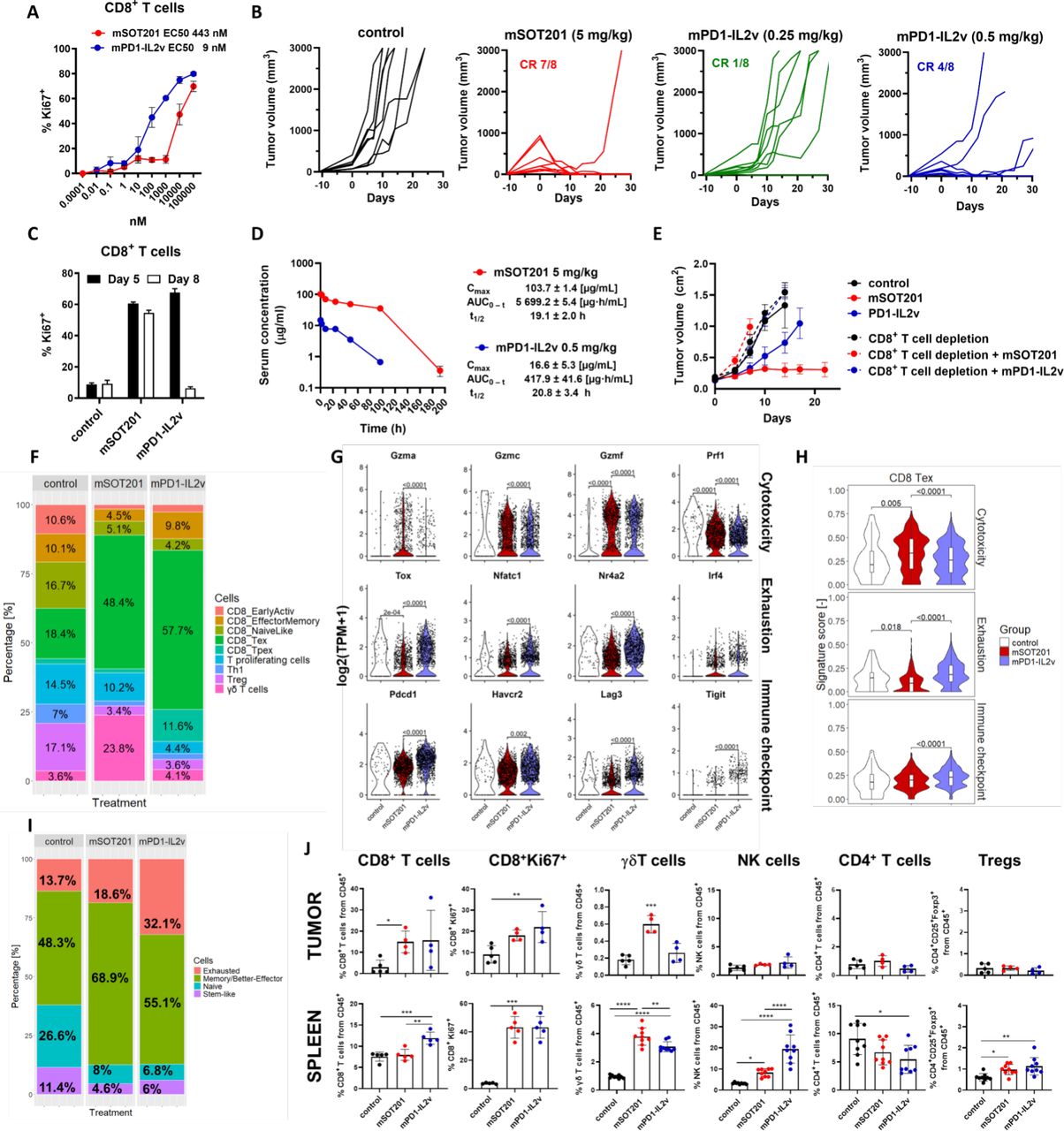
mSOT201 activation of CD8+ Tex cells and antitumor efficacy was compared with cis-acting IL-2/15Rβγ agonist mPD1-IL2v. mPD1-IL2v comprises the same anti-PD-1 mAb as mSOT201, but it is genetically fused with an IL-2 variant with abolished IL-2Rα binding via introduction of distinct mutations (IL2v) (online supplemental table 1). Unlike RLI-15m used in mSOT201, IL2v signaling via IL-2/15Rβγ is not attenuated. mPD1-IL2v induced a splenic CD8+ T-cell proliferation in vitro with~50-fold lower EC50 (9 nM) than mSOT201 (EC50 443 nM) (figure 4A). To compare their activity in vivo, non-equimolar doses of mSOT201 and mPD1-IL2v were selected based on a comparable activation of peripheral CD8+ T cells induced in vivo (figure 4C) and a maximum tolerated dose of each compound in the MC38 mouse tumor model (online supplemental figure 3). Selected efficacious doses were safe and no increase in biochemical parameters or serum cytokines was observed (data not shown). Surprisingly, mPD1-IL2v displayed a qualitatively less efficient antitumor efficacy at 0.5 mg/kg (3.05 µM) (4 complete responses (CR)/8) and 0.25 mg/kg (1.5 µM) (1 CR/8) leading to a high rate of tumor regrowth in comparison to mSOT201 at 5 mg/kg (30.5 µM) (7 CR/8) (figure 4B). mPD1-IL2v at 0.25 mg/kg and mSOT201 at 5 mg/kg stimulated a comparable splenic CD8+ T cell proliferation in vivo at day 5 but not at day 8 (figure 4C) pointing to pharmacokinetic differences between the compounds (figure 4D). The antitumor activity of mSOT201 and mPD1-IL2v was completely abolished by adding a CD8+ cell-depleting anti-CD8 mAb (figure 4E) suggesting that the quality of CD8+ T cells may account for differences in antitumor efficacy between both agonists. In accordance with scRNA-seq results in figure 3 mSOT201 expanded CD8+ Tex cells (48.4%), but at a lower extent than mPD1-IL2v (57.7%) (figure 4F). Furthermore, mPD1-IL2v markedly increased Tpex cells (11.6%) in comparison to mSOT201 (1.4%) and the control (1.9%). Unlike mSOT201, mPD1-IL2v did not drive the γδ T cell expansion. A similar decrease in Th1, Treg, CD8+ naïve-like and CD8+ early active cell populations was observed on treatment with both immunocytokines in comparison to the control (figure 4F). mSOT201 induced a higher expression of cytotoxic genes while showing lower gene expression for immune checkpoints and exhaustion in CD8+ Tex cells in comparison to mPD1-IL2v (figure 4G and H). These observations were confirmed by applying an additional scRNA-seq clustering according to10 (figure 4I, online supplemental figure 4). mPD1-IL2v increased exhausted T cells (32.1%) and decreased better/memory effector CD8+ T cells (55.1%) in comparison to mSOT201 which yielded 18.6% and 68.9%, respectively. Furthermore, mPD1-IL2v displayed lower cytotoxicity and higher checkpoint/exhaustion transcriptional profiles in both functional T cell types (online supplemental figure 4). Flow cytometry phenotyping did not reveal differences in relative abundance of CD8+ T, NK, CD4+ T and Treg cells in tumors (figure 4J). However, there was a significantly higher CD8+ T and NK cell expansion in spleens on mPD1-IL2v treatment, which suggests a higher peripheral sink. Notably, unlike mSOT201, mPD1-IL2v did not expand γδ T cells in tumors, although these cells showed a small increase in spleen. Both compounds similarly increased Treg cells in spleens (figure 4J). In conclusion, mSOT201 with an attenuated IL-15 showed a higher rate of complete tumor regressions in MC38 mouse tumor models with qualitatively superior CD8+ Tex cell revival displaying higher cytotoxicity and a lower exhaustion/immune checkpoints profile in comparison to a non-attenuated (with respect to IL-2/15Rβγ) mPD1-IL2v when compared at safe and efficacious doses. In contrast to mSOT201, mPD1-IL2v did not induce the expansion of γδ T cells in the tumor.

mSOT201 induces better effector CD8+ Tex driving a superior antitumor response compared with mPD1-IL2v. (A) mSOT201 and mPD1-IL2v-induced splenic CD8+ T-cell proliferation after 5 days in vitro. (B) Antitumor efficacy in MC38 mouse tumor model. (C) Splenic CD8+ T-cell proliferation in vivo after 5 and 8 days after injection of mSOT201 (5 mg/kg intravenous) and mPD1-IL2v (0.25 mg/kg intravenous). (D) Pharmacokinetic profile in vivo. (E) Antitumor efficacy in MC38 mouse model±CD8+ T-cell depletion (F) T-cell populations (scRNA-seq) in MC38 tumors 5 days after treatment with mSOT201 (5 mg/kg intravenous) or mPD1-IL2v (0.5 mg/kg intravenous). (G) Violin plots of selected genes in CD8+ Tex. (H) Violin gene signature plots in CD8+ Tex. (I) Functional T cells (scRNA-seq) using an alternative clustering according to.10 (J) Flow cytometry phenotyping of MC38 tumors and spleen day 5 post-treatment. Means±SEM. NK, natural killer; PD-1, programmed cell death 1; scRNA-seq, single-cell RNA sequencing; Tex, exhausted T cells; Treg, regulatory T cell.
mSOT201 promotes memory formation in OVA-primed CD8+ T cells, while mPD1-IL2v expansion is limited by peripheral sink
mSOT201 was the only treatment modality which significantly increased the relative number of the immunodominant RPL18-specific CD8+ T cells in MC38 tumors41 in comparison to the control after 5 days (figure 5A). In the spleen, these T cells were significantly increased on mSOT201 and mPD1-IL2v treatment in comparison to all other treatments (figure 5A). This supports the observation that differences in antitumor efficacy between IL-2/15Rβγ agonists mSOT201 and mPD1-IL2v are primarily driven by CD8+ T-cell quality rather than quantity. To further investigate this finding, the expansion of adoptively transferred naïve OVA-specific CD8+ T cells (OT-I) by mSOT201 and mPD1-IL2v on OVA priming in vivo was examined (figure 4B). mSOT201 expanded OVA-primed OT-I CD8+ T cells more potently than mPD1-IL2v after 5 days in vivo (figure 5C and D). Both mSOT201 and mPD1-IL2v expanded these cells only on OVA priming proving that T-cell receptor-induced activation of CD8+ T cells is a prerequisite for the stimulatory activity of these immunocytokines. The mSOT201 stimulatory effect was dependent on cis-activation as hPD1-mSOT201 was ineffective (figure 5C). Most importantly, mSOT201 did not expand endogenous (ie, not OVA-specific) CD8+ T cells in contrast to mPD1-IL2v showing that mPD1-IL2v suffers from the peripheral sink on antigen-irrelevant polyclonal CD8+ T cells (figure 5C and D). The establishment of the memory CD8+ T-cell population was investigated after 47 days (figure 5B and C). A lower number of OVA-specific memory CD8+ T cells was detected with mPD1-IL2v than with mSOT201 (figure 5E); however, both compounds induced functional memory T cells (figure 5C). As expected, neither hPD1-mSOT201 nor OVA alone established detectable memory CD8+ T cells (figure 5C) confirming that cis-activation via PD-1 expressed on OVA-primed CD8+ T cells together with IL-2/15Rβγ signaling is crucial. No expansion of endogenous polyclonal CD8 + T cells was observed with either treatment (figure 5E). In conclusion, IL-2/15Rβγ-binding attenuated mSOT201 drives a more potent expansion of antigen-primed CD8+ T cells through cis-activation, promoting their differentiation into memory T cells, compared with mPD1-IL2v, which is not attenuated for IL-2/15Rβγ binding. Lower expansion on mPD1-IL2v seems to be due to the high level of peripheral sink on antigen non-specific polyclonal CD8+ T cells due to its higher affinity to IL-2/15Rβγ.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
mSOT201 promotes memory formation in OVA-primed CD8+ T cells, while mPD1-IL2v expansion is limited by peripheral sink. (A) The RLP18-antigen specific CD8+ T cells from MC38 TILs and spleen 5 days after treatment. (B) Scheme of OT-I adoptive transfer. (C) Representative dotplots. (C, D) Quantification of OVA-specific CD8+ T cells (Ly5.1+) and endogenous CD8+ T-cell expansion on (C) day 5 and (D) day 47. All treatments were 5 mg/kg intravenous, except mPD1-IL2v was 0.5 mg/kg, intravenous. FACS, Fluorescence-Activated Cell Sorting; IL, interleukin; i.p, intraperitoneal; PD-1, programmed cell death 1; TILs, tumor-infiltrating lymphocytes.
Discussion
In this study, we describe a novel, affinity-optimized immunocytokine SOT201 based on an anti-PD-1 mAb fused to an attenuated IL-15/IL-15Rα sushi+ conjugate, which binds in cis to PD-1+CD8+ T cells and delivers IL-15 signaling via the IL-2/15Rβγ in vitro and in vivo. Moreover, we compared the functional composition of exhausted CD8+ TILs in MC38 tumors treated with mSOT201 and a 50-fold more potent mPD1-IL2v, showing that the strength of IL-2/15Rβγ signaling seems to determine the quality of the CD8+ T cell-mediated antitumor responses. We further describe a significant increase in γδ T cells on treatment with mSOT201 in contrast to mPD1-IL2v. Our data shows that SOT201 represents a promising IL-15-based candidate for efficient cancer immunotherapy.
The cis-activation, that is, binding of both PD-1 and IL-2/15Rβγ on the same cell, of human PD-1+CD8+ T cells with SOT201 was confirmed in vitro and in vivo. For NK cells, the cis-activation via PD-1 might not be as relevant as the expression of the IL-2/15Rβγ outnumbers the PD-1 molecules on the NK cell surface15 leading to a direct signaling via IL-2/15Rβγ independent of PD-1 binding. The PD-1-dependent stimulation of PD-1+CD8+ T cells was detected at~0.01 nM SOT201, a concentration that could be reached in tumors of patients on a systemic administration of tolerable doses. Docking to PD-1 by anti-PD-1 mAbs can reverse T-cell inhibition, although at higher concentrations (>1 nM) than those required for SOT201-induced T-cell activation. It has been documented that typically only~0.01% of systemically injected antibody reaches the tumor.42 However, the accumulation of an IL2v-based immunocytokine dosed at lower levels than typical antibody dosing was still detected in tumors of patients with cancer for up to 8 days, specifically in target antigen-positive tumor lesions.43 PD-1 expression on CD8+ TILs is significantly higher in comparison to CD8+ T cells in the periphery or non-tumoral tissues.11 44 mSOT201-induced cis-activation of PD-1+CD8+ T cells was crucial for the antitumor efficacy observed in all anti-PD-1 sensitive and resistant tumor models. This corroborates the preclinical findings of others using either IL-15-based or IL-2-based immunocytokines, where these responses were superior to CPI monotherapy alone, or to the combination of the cytokine and the CPI.10–15 SOT201 revived partially exhausted human T cells in vitro, aligning with the enhanced activation and cytotoxicity observed in primary human cancer TILs on stimulation with an attenuated IL-15-based PD-1-targeted immunocytokine.11
scRNA-seq analyses of CD8+ TILs from MC38 tumors revealed that similarly to anti-mPD-1 mAb treatment, the non-attenuated mPD1-IL2v expanded the progenitor CD8+ Tpex and CD8+ effector memory together with CD8+ Tex cells as similarly reported by others,10 12 which is distinct to mSOT201 treatment where only CD8+ Tex cells were the major expanded population. As IL-15 has been described to expand the CD8+ Tpex population from CD8+ TILs of patients with renal cell carcinoma ex vivo,45 it remains to be determined if mSOT201 differentiates these cells into CD8+ Tex with faster kinetics before the day of our analyses or if mSOT201 exclusively reinvigorates the CD8+ Tex population. The latter has been described so far only for a half-life extended IL-10-Fc fusion cytokine.46 IL-10-Fc was shown to directly enhance CD8+ Tex expansion and effector function by promoting their oxidative phosphorylation and metabolic reprogramming.46 Although an enhanced oxidative phosphorylation gene signature in CD8+ Tex cells on mSOT201 treatment was detected (data not shown), it remains to be further investigated if mSOT201 induces a similar metabolic reprogramming.
Importantly, this study showed that cytokine attenuation in a cis-acting PD-1-targeted IL-15-based immunocytokine offers an advantage in reinvigorating highly cytotoxic, less exhausted CD8+ Tex cells in tumors. This effect was superior to that of the 50-fold more potent cytokine non-attenuated IL-2/15Rβγ agonist mPD1-IL2v, and appears to underlie the higher rate of durable antitumor responses in the MC38 mouse model, when compounds were injected at safe and efficacious doses. The MC38 colorectal cancer model used in our study is a well-established model with a high tumor mutational burden associated with high immunogenicity and considerable responsiveness to CPI treatment.41 No substantial quantitative differences were detected in the expansion of the dominant neoantigen-specific CD8+ T cells between the compounds. In contrast to mSOT201, mPD1-IL2v substantially activated antigen-irrelevant polyclonal CD8+ T cells. This suggests that the exposure of the tumor microenvironment to mPD1-IL2v might be limited due to high target-mediated drug disposition in the periphery. Thus, various immune cells expressing IL-2/15Rβγ within the body represent a sink for mPD1-IL2v independent of PD-1-binding, but not for mSOT201 enabling superior stimulation of PD-1+CD8+ T cells within the tumor on mSOT201 treatment. Consequently, the stimulatory activity of mPD1-IL2v is less dependent on PD-1-mediated cis-interaction, as the IL2v affinity to IL-2/15Rβγ outweighs its PD-1 binding. This leads to more profound expansion of NK cells in the spleen on mPD1-IL2v treatment compared with mSOT201, which shows a strong avidity effect for PD-1 and IL-2/15Rβγ positive cells. IL-2 and IL-15 can display biological differences which are determined by interaction with their specific IL-2Rα and IL-15Rα and their spatiotemporal action.1–4 IL-2 is secreted by cells and binds with high affinity to IL-2Rαβγ, whereas IL-15 is mainly membrane-bound to its IL-15Rα on the cell surface, where it forms a complex that is trans-presented with high affinity to mid-affinity IL-2/15Rβγ. Therefore, the phenotypic differences induced by IL-2 and IL-15 in CD8+ T cells are likely not a result of distinct signal transduction in cells from the shared IL-2/15Rβγ, but rather a result of a stronger and more persistent signaling of IL-15Rα/IL-15 complex compared with stimulation with IL-2.2 However, there is no information about similar biological differences between mutant IL-2 and IL-15 cytokines and even less in a cis-targeting antibody fusion which likely affects the duration of the signaling via IL-2/15Rβγ. Here, no receptor α-chain binding occurs for mPD1-IL2v whereas the RLI-15m used in mSOT201 bears the sushi+ domain of the IL-15Rα, which substantially increases the affinity to the IL-2/IL-15Rβγ compared with the respective IL-15 mutant with reduced IL-2/15Rβγ binding. Therefore, both compounds mSOT201 and mPD1-IL2v signal via the same IL-2/15Rβγ but with different affinities and duration of the signaling. It seems that for mSOT201 there is an optimally balanced affinity-based strength and duration of signaling via the IL-2/15Rβγ and a favorable pharmacokinetic profile that limits the peripheral CD8+ T-cell sink in vivo resulting in a qualitatively superior antitumor response over mPD1-IL2v.
Another novel observation of our study is the substantial increase in the γδ T cells after mSOT201 administration in MC38 tumors, which were not observed with mPD1-IL2v or other immunocytokine treatments.10–15 PD-1-targeted IL-2/15-based immunocytokines acting on γδ T cells have not been characterized so far. In our study, mSOT201 drove γδ T-cell expansion with a higher potency compared with mPD1-IL2v in the spleen. This expansion was PD-1 dependent, which is in line with the moderate levels of PD-1 together with elevated IL-2/15Rβγ levels in γδ T cells compared with αβ T cells. Moreover, IL-2 and IL-15 are the main inducers of cytotoxic γδ T cells.47 γδ T cells may at least mechanistically represent a target population for mSOT201 in our study, even though the antitumor activity in the MC38 mouse tumor model seems to be largely driven by CD8+ T cells. The role of mSOT201-expanded γδ T cells is not clear in this model, although IFN-γ producing γδ T cells were shown to support CD8+ T cells48 and antitumor responses to CPI in colorectal cancer.49 It cannot be ruled out that the ability of mSOT201 to strongly reinvigorate the antitumor response in different models may be in part dependent on the supportive action of γδ T cells in addition to CD8+ T cells. This may be particularly relevant for several cancer types where increased γδ T cells were shown to correlate with a positive outcome in patients.50
Preventing or reverting an exhaustion/dysfunctional state of tumor-antigen specific CD8+ T cells as well as their expansion is one of the major goals in cancer immunotherapies. We conclude that SOT201 represents a promising therapeutic candidate directed preferentially to the PD-1+ TILs with a balanced cytokine activity and potency to revive better effector CD8+ Tex cells in tumors. SOT201 is currently being evaluated in the Phase I clinical study VICTORIA-01 (NCT06163391) in patients with metastatic advanced cancer.
Data availability statement
Data are available upon reasonable request. Data are proprietary of SOTIO Biotech AG and available on reasonable request. The datasets generated and/or analysed during the current study are available from the corresponding authors on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors would like to acknowledge the supportive work of Ondrej Novotny, Renata Tureckova and Pavlina Jungrova. The authors would like to thank Christoph Rehfuess and Anna Jirovec for their valuable contribution to the manuscript review process.
References
Footnotes
Contributors IA is the guarantor of the study. IA, HM, LPJ, MR, DB, UM, MK, RS and MS designed and developed the study. PM, KD, IM, KHr, DG, OM, MF, NP, KHl and ZA performed the in vitro experiments, aided in data collection and performed the analyses. HM, PM, VM, LK, PD, KB, KS, ZA, MS, KHl and RM performed the in vivo and ex vivo experiments, aided in data collection and performed the analyses. ES performed scRNA-seq evaluation and analyses. IA, HM, PM and MS did the formal analyses and interpreted the data. IA and MS wrote the original draft. All other authors contributed to the writing and review of the manuscript. The corresponding authors had final responsibility for the decision to submit for publication on behalf of the collaborative authors’ group. All authors were not precluded from accessing data in the study, and they accept responsibility to submit for publication.
Funding This work was supported by SOTIO Biotech AG, by the grant of Institute of Molecular Genetics of the Czech Academy of Sciences No. RVO 68378050, by the project National Institute for Cancer Research (Programme EXCELES, ID Project No. LX22NPO5102) - Funded by the European Union – Next Generation EU and by the institutional research concept of the Institute of Microbiology of the Czech Academy of Sciences (RVO 6138897).
Competing interests All authors except VM, KB, MS, RM, MR and MK are employees of SOTIO Biotech a.s and DB is the CEO of Cytune Pharma.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.