Article Text

Abstract
Background Curative responses to immunotherapy require the generation of robust systemic immunity with limited toxicity. Recruitment of T cell populations such as precursor exhausted T cells (Tpex) from lymphoid tissues to tumors is a hallmark of effective treatment. However, the ability to efficiently induce this recruitment is lacking in current immunotherapy approaches. Furthermore, systemic administration of immunotherapies frequently results in dose-limiting toxicities, yielding an inadequate therapeutic window for eliciting durable responses.
Methods In this investigation, we evaluated the safety and antitumor efficacy of locally administered interleukin 12 (IL-12) using a clinically translatable cytokine delivery platform (NCT05538624) to identify Tpex recruitment capabilities at tolerable cytokine doses.
Results We show IL-12 cytokine factories can effectively treat a broad spectrum of cancer types. Single-cell RNA sequencing data suggests that the antitumor efficacy seen in our studies was due to retinal pigmented epithelial cells-mIL12 treatment inducing differentiation of Tpex cells within the tumor microenvironment. When administered in combination with checkpoint therapy, IL-12 cytokine factory treatment generated systemic abscopal immunity, preventing subcutaneous tumor outgrowth in 8/9 mice with colorectal cancer and lung metastasis in mice with melanoma. Furthermore, this platform was well tolerated in a non-human primate without signs of toxicity.
Conclusions Our new immunotherapy approach provides a robust strategy for inducing Tpex recruitment and systemic immunity against a range of solid peritoneal malignancies, many incurable with current immunotherapy strategies. Notably, these features were achieved using IL-12, and by leveraging our technology, we avoided the toxicities that have prevented the translation of IL-12 to the clinic. Our findings provide a strong rationale for the clinical development of IL-12 cytokine factories.
- Cytokine
- Abscopal
- Colorectal Cancer
- Gastric Cancer
- Immune modulatory
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Previously, we have shown that utilization of our cytokine factory technology for local delivery of interleukin (IL)-2 is well tolerated and highly efficacious, motivating the initiation of a phase I clinical trial in patients with ovarian cancer (NCT05538624).
WHAT THIS STUDY ADDS
Here, we show that delivery of the cytokine IL-12 using the same technology is capable of eradicating local and distal tumors in a variety of aggressive cancer models. Additionally, we demonstrate that the level of efficacy achieved appears to be due to the diverse immune response generated, including the induction of precursor exhausted T cell recruitment to the tumor.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our results provide a strong rationale for the treatment of humans with IL-12 producing cytokine factories for the treatment of cancer.
Background
While immunotherapy represents the most important advance in the field of oncology in many decades, the majority of patients with cancer fail to experience durable responses from these treatments, and many cancer types, such as pancreatic cancer, rarely exhibit any response to immunotherapies. In the context of solid organ malignancies, these shortcomings are frequently a consequence of poor recruitment, infiltration, or retention of lymphocytes or the induction of dysfunctional T cell states following chronic antigen exposure.1 2 Furthermore, systemic administration of immunotherapies can lead to life-threatening toxicities, greatly limiting the therapeutic window.3–5 Development of an effective immunotherapy for treatment of solid tumors requires a technology capable of overcoming these challenges to (1) successfully generate a systemic antitumor immune response, (2) generate a durable response unlikely to enter a dysfunctional state, and (3) limit systemic immunotherapy exposure to increase tolerability.
Effective antitumor immune responses to immunotherapy require immunity activation at extratumorous sites.6 7 Mouse and human studies evaluating T cell repertoires in blood and tumors before and following treatment have implicated clonal replacement and the continual recruitment of T cells in responders.8–13 Furthermore, pharmacological or surgical inhibition of lymphocyte egress from lymph nodes (LNs) abrogates immunotherapy efficacy, whereas the delivery of checkpoint inhibitors directly to LNs enhances their effects.6 7 14 15 Indeed, the recent clinical successes of neoadjuvant checkpoint inhibitor treatment in locally advanced resectable solid tumors suggest that activating antitumor immunity within LNs that have seen tumor antigens is essential for generating systemic immune responses and improving outcomes following surgery.16–18
In addition to the successful activation of the immune system, effective immunotherapy must also induce a durable antitumor response. T cell exhaustion, a dysfunctional state of T cells characterized by a lack of responsiveness resulting from chronic antigen stimulation, is now appreciated as a primary driver of therapeutic resistance.19–22 Nonetheless, there is an emerging appreciation for precursor populations of these terminally exhausted T cells that are less differentiated and are essential mediators of effective responses to checkpoint inhibitor therapy. These progenitor or precursor exhausted T cells (Tpex) exhibit a range of stem-like features, sharing many traits with memory T cell populations.23 Tpex cells express the transcription factor TCF-1 and accumulate in the tumors, driving responses to immune checkpoint blockade and vaccination.21 24–31 Consistent with the identification of LNs as orchestrators of antitumor immunity, evidence in mice and humans suggests that a pool of renewing Tpex resides within LNs and continuously migrates to tumors where they may be further activated by dendritic cells to elicit antitumor immunity.11 32–37 Thus, the generation, maintenance, and continual recruitment of tumor-specific Tpex and diverse T cell populations along the exhaustion spectrum represent an essential feature of effective immunotherapy.
While systemic administration of immunotherapies has the potential to generate curative responses through the generation of antitumor immunity throughout the patient, such approaches harbor the potential to elicit lethal toxicities. Systemic cytokine therapy (eg, interleukin (IL)-2) can induce cytokine release syndrome, and cell therapies such as chimeric antigen receptor T cells can additionally induce tumor lysis syndrome or immune effector cell-associated neurotoxicity syndrome. In addition, immune checkpoint blockade can induce a range of autoimmune immune-related adverse events, all of which limit the therapeutic window.3–5 These dose-limiting toxicities often prevent the administration of the therapies at levels necessary for eliciting responses.
Recently, we developed a cytokine delivery platform capable of producing high concentrations of immunotherapies at the sites of tumors while maintaining negligible levels in the plasma of patients. In this manuscript, we refer to this platform as “cell factories”. This technology is currently undergoing clinical trials for the treatment of ovarian cancer (NCT05538624).38 Here, we sought to leverage this technology to generate durable, systemic antitumor immunity capable of continuously recruiting Tpex populations and eliminating local and metastatic tumors. By screening various cytokine payloads, we identify ideal implementations capable of recruiting enhanced T cell diversity and curative responses while maintaining high tolerability to treatment. After identifying cell factories with these features, we validate that delivery of these cytokines indeed translates to safe and efficacious treatment through testing against various mouse cancer models, including metastatic melanoma, pancreatic cancer, and colorectal cancer.
Methods
Cell culture and engineering
Cell culture media and associated reagents were purchased through Fisher Scientific. Transfection reagents (lipofectamine 3000) and selection media (puromycin) were purchased from Invitrogen. All cell lines tested negative for mycoplasma contamination. All cell lines used in our studies were authenticated by the vendor.
ARPE-19 cells (American Type Culture Collection (ATCC)), MC38 cells (Kerafast) and B16LN6 cells (LN6-987AL) melanoma cells (generated through serial in vivo passaging from LN metastases, described previously, (41)) were cultured using Dulbecco’s Modified Eagle Medium (DMEM/F-12), with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (AA). KPC (CancerTools.org),39 and Pan02 cells (Divison of Cancer Treatment and Diagnosis (DCTD) Tumor Repository, National Cancer Institute) were cultured using Roswell Park Memorial Institute 1640, 10% FBS, and 1% AA. The media was changed three times weekly. Human ARPE-19 cells (lot #70022669) have been characterized by ATCC by cytogenetic analysis to be diploid and have the following short tandem repeat profile: Amelogenin: X,Y; CSF1PO: 11; D13S317: 11,12; D16S539: 9,11; D5S818: 13; D7S820: 9,11; TH01: 6,9.3; TPOX: 9,11; and vWA: 16,19.
Cell transfection/transduction
ARPE-19 cells (ATCC) were engineered to express cytokines of interest (mIL12, mIL2, mIFNg). Expression vectors and helper plasmids were designed and purchased through VectorBuilder. KPC and Pan02 cells were engineered to express firefly luciferase. Cells were transfected or transduced as described previously.38 40
Core shell cell encapsulation
Capsules were generated as described previously.38 40 Briefly, alginate was dissolved at 1.4% w/v in saline and sterile filtered. Cells were resuspended in alginate at a concentration of 10.5×106 cells/mL. Cells were encapsulated using a custom-built, two-fluid co-axial electrostatic spraying device. Alginate droplets were expelled from a co-axial needle into barium chloride crosslinking solution. They were subsequently washed with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer and maintained with normal cell culture techniques.
Cell viability post encapsulation
Following encapsulation, a subset of capsules were washed with 5 mL Dulbecco’s Phosphate-Buffered Saline (DPBS) and stained using a stock 2 µM calcein AM and 4 µM EthD-1 in complete media. The sample was incubated for 20 min and imaged using a fluorescence microscope.
ELISA
An individual capsule was added to a 96-well plate (n=6–8) in 200 µl for 24 hours at 37 degrees in a 5% CO2 humidified incubator. Cell supernatant was collected from each well and assayed via ELISA according to manufacturer protocols. Kits were obtained commercially from R&D Systems. All samples were run in duplicate.
Animal studies
Mouse studies
C57BL/6 mice (Charles River Laboratories, mixed gender), aged 8–10 weeks were used for in vivo studies. All animal experiments were approved by Rice University’s Institution Animal Care and Use Committee (IACUC). All biological samples implanted into animals were approved by Rice University’s Institutional Biosafety Committee. For intraperitoneal (IP) tumor models of MC38 colorectal tumors; 1×106 cells suspended in Hanks’ Balanced Salt Solution (HBSS) were IP injected into the lower right abdomen. For IP tumor models; 1×105-5×106 cells suspended in HBSS were IP injected into the lower right abdomen. For tumor models of metastatic melanoma tumors in the lungs; 2×105 cells suspended in HBSS were injected into the lateral tail veins 4 days following IP implantation of 105 cells. The approximate dose of each cytokine administered from the cytokine factories was as follows: ∼2.5 µg/day for retinal pigmented epithelial cells (RPE)-mIL2, ∼1.0 µg/day from RPE-mIL12, and∼0.5 µg/day for RPE-mIFNγ. Cytokine production was calculated using ELISA. Tumors were injected and allowed to develop in vivo for 1 week before treatment. For all studies using In Vitro Imaging System (IVIS) imaging for tumor growth tracking, mice were imaged and stratified into treatment groups 1 day prior to surgery using the methods described in the IVIS imaging section below. After stratification for tumor size, animals were randomly assigned to treatment groups. For tumor measurements, experimenters were not blinded (as is commonly accepted in the field). Antitumor efficacy of therapy was confirmed by multiple investigators.
Orthotopic injection of pancreatic cancer cells in mice
Mice were sedated and anesthetized in accordance with approved animal protocols at Rice University. A surgical blade (15T; Sklar) was then used to cut a 0.5–0.75 cm incision through the skin and muscle on the upper right torso below the ribcage. Cells were resuspended at a 1:1 ratio in Geltrex basement membrane matrix (A1413202, Thermo Fisher Scientific) and administered via injection into the pancreas. The abdominal muscle was closed by suturing with 5–0 Ethicon black Polydioxanone (PDS)-absorbable or other 5.0–6.0 monofilament absorbable sutures. The external skin layer was closed with PDS suture as previously described.
Experimental controls
Sham: all mice given sham surgery received IP surgery and were administered 1 mL sterile saline. RPE: RPE capsules contained the same density of cells as experimental capsules but contained naïve cells. Anti-programmed cell death protein-1 (aPD1): all mice treated with aPD1 antibodies (29F.1A12, Bio X Cell) received IP injection of 200 µg per mouse at day 0, 3, 7, and 10 post cytokine factory treatment.
Subcutaneous tumor growth tracking
For subcutaneous MC38 tumor models, the tumor size was measured using a digital caliper and tumor volume was calculated using the formula V=0.5×(height)×(width2).
Immune cell depletion studies
For depletion studies, isotype control (LTF-2), anti-CD8a (2.43, Bio X Cell), anti-CSF1R (AFS98, Bio X Cell), anti-CD20 (MB20-11, Bio X Cell), anti-NK1.1 (PK136, Bio X Cell), or anti-CD4 (GK1.5, Bio X Cell) antibodies were administered via IP injection at a dose of 100 µg per animal at day −2, 0, and 2 post RPE-mIL12 implantation.
IP tumor growth tracking
Animals injected with Pan02-Fluc cells were imaged using IVIS 6 days after injection and stratified into experimental groups based on the luminescent signal. Imaging and implantation methods are expanded below.
IP surgical implantation of capsules in mice
Implantation studies were carried out as previously described.38 40 Briefly, mice were sedated and anesthetized in accordance with approved animal protocols at Rice University. A surgical blade (15T; Sklar) was then used to cut a 0.5–0.75 cm midline incision through the skin and the linea alba into the abdomen. Capsule implants were administered using sterile transfer pipets. The abdominal muscle was closed by suturing with 5–0 Ethicon black PDS-absorbable or other 5.0–6.0 monofilament absorbable sutures. The external skin layer was closed with PDS suture as previously described.
IVIS imaging
Mice were anesthetized in accordance with approved animal protocols at Rice University and injected in the IP space with D-luciferin (300 µg/mL, 200 µL; PerkinElmer). Animals were then transferred to the IVIS manifold (IVIS Spectrum, PerkinElmer) where they were kept under isoflurane anesthesia (0.25 L/min) and maintained warm on a heated stage. Photographs and luminescent images were acquired 10 min after injection. Luminescent exposures were set to 1 s with the binning set at medium, the excitation set to block, and the electron multiplying (EM) gain set to “off” with 0 s delays between acquisitions. Subsequent to stratification, the image of each mouse was individually cropped and stitched to create a collage of each treatment group.
Single-cell RNA-seq
Single-cell 5’ Gene Expression Library was prepared according to Chromium Next GEM Single Cell Immune Profiling Solution 5’v2 (10× Genomics) as described previously.41 The resulting libraries were sequenced on an Illumina NovaSeq 6000 flow cell. Transcripts within each cell were counted using the 10× Cell Ranger V.5.0.1 pipeline, with genome mapping using STAR V.2.7.2a. Multiplets were removed using DoubletFinder V.2.0.342 and AMULET V.1.1.43 To identify immune cell populations, gene expression data was visualized using Uniform Manifold Approximation and Projection (UMAP) embedding in Loupe Browser V.6.1.0. The cell identities of the resulting clusters were resolved using the following markers: Cd3e and Prf1 (T cells/natural killer (NK) cells), Cd19 (B cells), Csf1r and Fn1 (myeloid cells), and Ly6g and Hdc (granulocytes). The myeloid cluster was further separated into monocytes (Flt3−) and dendritic cells (Flt3+). On the basis of these markers, individual cell barcodes were assigned to their corresponding immune cell type in the Loupe Browser.
To assess changes in the infiltrate immune composition, changes in cell proportions were calculated between each implant’s surface topology for each immune cell type. The significance of these changes was calculated using Fisher’s exact test in R (V.3.6.1), and the resulting p values were adjusted using the Benjamini-Hochberg false-discovery rate correction method. NicheNet was used to model ligand-target interactions between different immune cell types.44
Primate studies
A non-human primate (NHP) (adult female Mauritian cynomolgus monkey; age, 12–14 years) was purchased from DSP Research Services. All animal procedures and postoperative care were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the UIC and approved by the IACUC of UIC.
IP surgical implantation of capsules in NHPs
Capsule delivery was performed under standard sterile precautions in a surgical suite as previously described.38 Briefly, the anterior abdomen was shaved and cleaned. Under general anesthesia, a small (2 cm) supraumbilical incision was created and a 5–12 mm trocar (Ethicon Endo-Surgery) was inserted. Pneumoperitoneum was created with CO2 at a pressure of 10–14 mmHg. Under the view of laparoscopy, another small incision (1 cm) was created; an additional 5–12 mm trocar was inserted into the peritoneal cavity for capsule administration. The capsule dose was administered in 50 mL of saline via a sterile 60 mL catheter tip syringe. Capsules were infused into the IP cavity via the trocar incision and distributed throughout the IP cavity space. The incisions were sutured by layers using Vicryl 3–0 suture (Ethicon) for the muscle and Vicryl 4–0 suture (Ethicon) for the skin (subcuticular).
Statistics
The sample size was predetermined from pilot experiments and/or experiments that have been done in the past, to obtain statistically significant data. Experiments were repeated at least once, or data were compiled from two independent experiments unless otherwise stated in the respective figure legend. Replicates were reproducible. All statistical analyses were conducted using GraphPad Prism V.9. One-way analysis of variance tests with the Holm-Sidak multiple comparisons methods were used to determine p values for flow cytometry datasets. Survival curves were analyzed using the log-rank Mantel-Cox test. Unless otherwise indicated as a replicate measurement, data were taken from distinct samples.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Results
Cytokine factories were designed to locally activate innate and adaptive immune cells
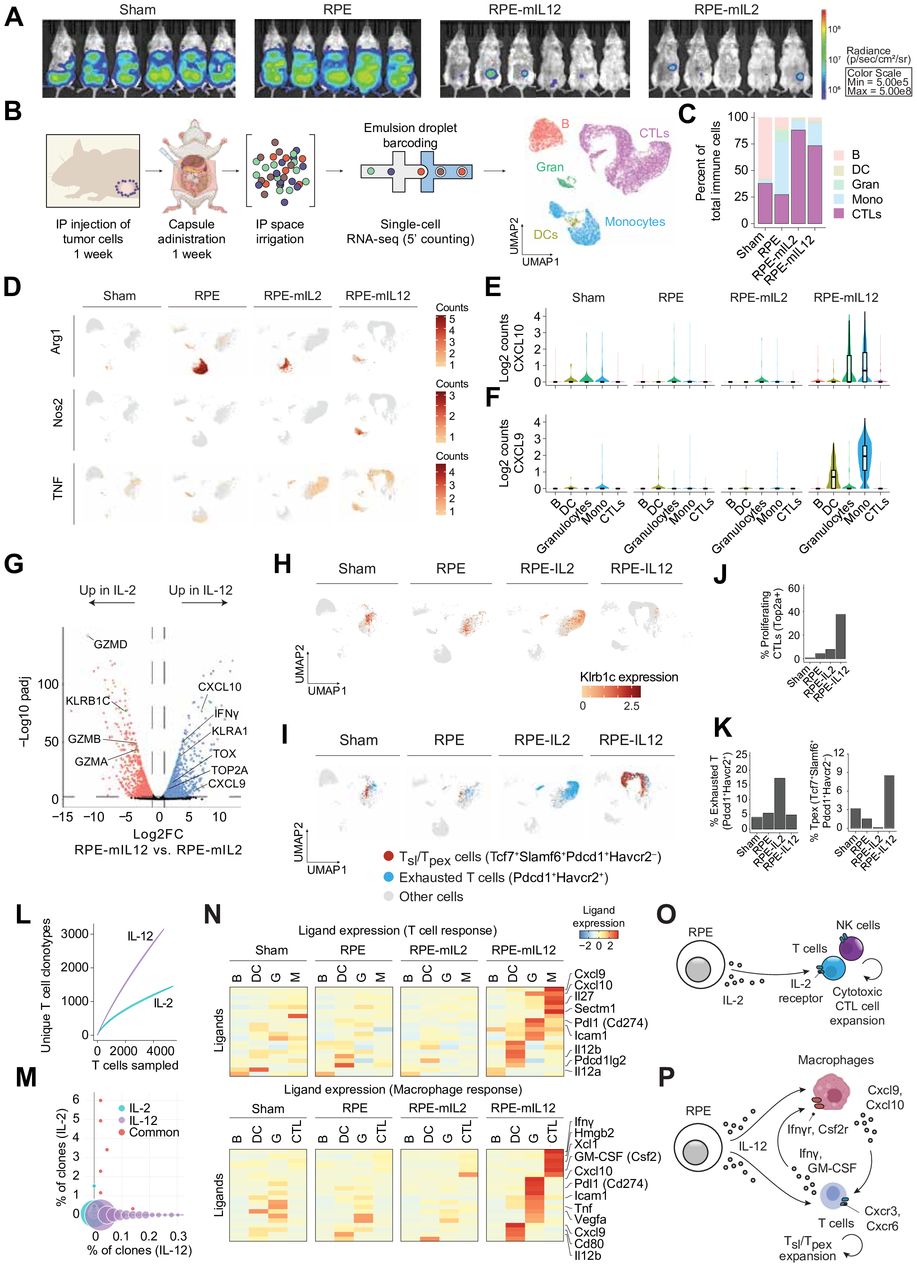
We fabricated proinflammatory cytokine factories as previously described.38 40 Briefly, RPE were genetically engineered to overexpress proinflammatory cytokines (IL-2, IL-12, or IFN-γ). The engineered cells were then encapsulated within porous inflammatory hydrogel (sodium alginate) microparticles. To ensure a consistent material-related effect study to study, the cell loading density (10.5 M cells per 1 mL of alginate) was maintained across all batches of cytokine factory produced. The amount of cytokine produced varies by cytokine. This composite system functions as a continuous source of cell-produced cytokines and can activate both innate (macrophage activation by alginate45) and adaptive (production of inflammatory cytokines) immune cells in vivo (figure 1A). Following cell engineering, encapsulation, and viability confirmation (figure 1B), the systems are referred to as RPE-mIL2, RPE-mIL12, or RPE-mIFNγ depending on the cytokine they were engineered to produce.

Cytokine factories activate innate and adaptive immune cells locally to elicit antitumor efficacy in mice. (A) Schematic detailing the components and variety of functions of cytokine factories. The schematic was created using biorender.com. (B) Darkfield images of cytokine factories prior to intraperitoneal implantation (dark-field and fluorescence (green fluorescent protein), 2×). Scale bars, 2 mm. (C) Tumor weight from individual mice 1 week after treatment with cytokine factories. Tumors were weighed using an analytical balance. (D) Representative macroscopic images of the melanoma tumors inside the abdominal cavity of animals from each treatment group. Images were taken immediately after sacrifice (day 7 post-treatment, day 14 post-tumor inoculation). (E–H) Cytokine concentration in the IP fluid (local concentration) of animals bearing abdominal metastatic melanoma tumors and treated with RPE-mIFNg, RPE-mIL12, RPE-mIL2, or sham surgery. Cytokine concentrations were determined using ELISA. (I–L) Cytokine concentration in the blood (systemic concentration) of animals bearing abdominal metastatic melanoma tumors and treated with RPE-mIFNg, RPE-mIL12, RPE-mIL2, or sham surgery. Cytokine concentrations were determined using ELISA. Data from C–L are representative of two independent experiments. IL, interleukin; IP, intraperitoneal; NK, natural killer; RPE, retinal pigmented epithelial cells.
Cytokine factories delay melanoma tumor growth in the IP cavity of mice
To rapidly screen cytokine factories for antitumor efficacy in vivo, we developed a mouse model of metastatic melanoma within the peritoneal cavity. While metastasis of cutaneous melanoma to the peritoneum is a rare event, it is extremely aggressive and uniformly incurable.46 This aggressive model spreads rapidly throughout the abdomen in mice (online supplemental figure S1) and recapitulates late-stage melanoma that has metastasized. Previously, we developed a mouse model of metastasis through serial in vivo passaging of the non-metastatic melanoma B16-F0 syngeneic in C57BL/6 mice. Mice were transplanted with B16-F0 parental cells, and LNs were harvested after 1 month of tumor growth. Tumor cells were isolated from any LNs bearing metastases and expanded into de novo cell lines, which were in turn transplanted into new mice. This process was repeated over six generations, resulting in B16LN6 cells, which metastasize to LNs readily and exhibit immunosuppressive features that enhance their metastatic potential.47
Supplemental material
Our developed model of metastatic melanoma is suitable for rapid screening of cytokine factories due to its aggressive nature and need for rapid treatment. Accordingly, we used this model to select highly effective and fast-acting cytokine factories. Briefly, 1.0e5 B16LN6 cells were injected into C57BL/6 mice (mixed gender). Tumors were left to develop for 7 days, and then animals were randomly divided into groups of 4 and treated with RPE-mIL2, RPE-mIL12, RPE-mIFNγ, RPE (control capsules with naïve, parental RPE cell line), or sham surgery. After a 7-day treatment (14 days after tumor inoculation), the animals were sacrificed, imaged, and the tumors were extracted from the IP cavity and weighed. RPE-mIL2, RPE-mIFNγ, and RPE-mIL12 treatment caused tumor reduction of greater than 50% (compared with sham mice) (figure 1C). RPE treatment did not show antitumor efficacy in this tumor model. Representative images of animals from each treatment group similarly illustrate the extent of tumor reduction resulting from RPE-mIL2, RPE-mIL12, and RPE-mIFNγ treatment (figure 1D). Thus, all three cytokine factories were selected for further investigation.
The local (IP fluid) and systemic (blood) cytokine concentrations were analyzed to assess the longevity of cytokine factory production and the extent of cytokine retention in the local cavity. Notably, for RPE-mIL2, RPE-mIL12, and RPE-mIFNγ, after 1 week, cytokine concentrations were high (greater than 2 ng/mL) and remained within the local fluid (figure 1E–H local, figure 1I–L systemic). These results suggest the broad utility of these factories for continuous local delivery of proinflammatory cytokines in vivo. Notably, RPE-mIL12 treatment resulted in an increased local concentration of mIL12 and IFN-γ, demonstrating bioactivity via its ability to induce the production of IFN-γ from the host immune system (figure 1F). Similarly, the measured concentration of mIL2 was higher than the amount administered, demonstrating its ability to interact with the host T cells and induce the production of supplemental IL248 (figure 1G). Since RPE-mIL12 treatment induced levels of IFN-γ similar to the RPE-mIFNγ treatment, the latter was deprioritized in subsequent experiments. Altogether, these results highlight the bioactivity of cell-generated cytokines and the potential for cytokine factories as a viable treatment for metastatic tumors.
RPE-mIL12 treatment induces Tpex formation to eliminate pancreatic tumors
We next sought to determine whether the IL2 or IL12 cytokine factories are capable of inducing features necessary for driving effective immunotherapy responses. Pancreatic cancer commonly metastasizes within the peritoneum, is the deadliest common malignancy, and is notoriously resistant to immunotherapy.49–51 Thus, to compare the extent of adaptive immune system activation and antitumor efficacy of RPE-mIL2 and RPE-mIL12 in a traditionally challenging setting, we used a mouse model of pancreatic cancer. Similar to the experimental design described above, we injected C57BL/6 mice (mixed sex) with 5e6 PAN02-fluc cells and allowed the tumor cells to disseminate for 1 week. The animals were imaged using IVIS to ensure the viability of tumor cells before treatment and to stratify mice into four treatment groups (sham, RPE, RPE-mIL2, and RPE-mIL12) based on similar extent of tumor burden before implantation. After 5 days of cytokine factory administration, the animals were imaged again to evaluate the extent of treatment-induced tumor cell death. Strikingly, RPE-mIL2 and RPE-mIL12 treated animals had significantly fewer viable tumor cells than control animals, highlighting the ability of cytokine factories to rapidly activate the immune system and induce antitumor effects (figure 2A, online supplemental figure S2). A week after treatment administration, the animals were sacrificed and cells from the IP cavity were collected and investigated using single-cell RNA sequencing. UMAP embedding revealed five major clusters well represented by all four conditions (figure 2B and online supplemental figure S3A). The clusters were classified by immune cell type using standard cell type-specific markers (figure 2B). Cluster identities were also validated by determining the top differential genes expressed by each population (online supplemental figure S3B).

RPE-mIL12 treatment induces Tpex formation to eliminate pancreatic tumors. (A) Luminescent images of mice with pancreatic cancer (n=5–6/group) 5 days after treatment with RPE, sham, RPE-mIL2, or RPE-mIL12. (B) Workflow to isolate cells for scRNA-seq and to process the resulting data. Immune cells were collected from the space surrounding IP tumors 2 weeks after administration of RPE capsule control, or RPE-IL2 or RPE-IL12 capsules. (C) Composition of immune cells recruited to the tumor site at 2 weeks, as classified in A. (D) Expression of individual monocyte/macrophage-related genes across conditions. Expression of CTL-recruiting chemokines (E) CXCL10 and (F) CXCL9 across cell types. (G) Gene expression changes in CTLs recruited to RPE-IL12 versus RPE-IL2-treated tumors. (H) Expression of Klrb1c across conditions. (I) Tpex cells (Tcf7+Slamf6+Pdcd1+Havcr2−) and exhausted T cells (Pdcd1+Havcr2+) across conditions. Percent of total CTLs that are (J) proliferative (Top2A+) or (K) Tpex and exhausted cells across conditions. (L) Number of unique TCR clonotypes versus total T cells isolated from tumors treated with RPE-IL2 and RPE-IL12 capsules. (M) Frequency and overlap of TCR clonotypes present at tumors treated with RPE-IL2 or RPE-IL12 capsules. (N) NicheNet analysis of the expression of CTL-interacting and macrophage-interacting ligands enriched in RPE-IL12 across immune cell types. (O) Model of RPE-IL2-mediated activation of T and NK CTLs. (P) Model of RPE-IL12-mediated activation of inflammatory macrophages and Tpex cells. CTLs, cytotoxic T lymphocytes; DCs, dendritic cells; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; IP, intraperitoneal; NK, natural killer; RPE, retinal pigmented epithelial cells; scRNA-seq; single-cell RNA-seq; TCR, T cell receptor; Tpex, precursor exhausted T cells; UMAP, Uniform Manifold Approximation and Projection.
The administration of RPE capsules clearly affected the composition of the immune infiltrate present at the tumor sites (figure 2C). Engraftment of the RPE capsule control alone resulted in the accumulation of monocytes, while IL-2 and IL-12-secreting capsules recruited cytotoxic lymphocytes (CTLs; T and NK cells) to the tumor microenvironment (TME). Monocytes trafficked to the capsule control and RPE-mIL2 capsules exhibited an M2-like immunosuppressive phenotype with high Arg1 expression, consistent with previous findings that IL-2 capsules become fibrosed over time40 (figure 2D). In contrast, RPE-mIL12 capsules primarily recruit inflammatory M1-like monocytes with high Nos2 expression (figure 2D). The inflammatory monocytes in RPE-IL12 also secrete high levels of CXCL9 and CXCL10 (figure 2E,F), chemokines that play a critical role in T-cell recruitment during immunotherapy.52
RPE-mIL2 and RPE-mIL12 capsules each recruited a unique population of CTLs to the tumor, thereby promoting a different type of antitumor response (figure 2G). We found RPE-mIL2 capsules to primarily activate NK cells, as evidenced by the enrichment of NK cell-related genes within the CTL population, including Klrb1c and granzymes (figure 2H, online supplemental figure S3D). In contrast, RPE-mIL12 capsules recruit and activate T cells and promote the generation of ‘precursor exhausted’ T cells (Tpex) that have been associated with response to immunotherapy53 (figure 2I–K and online supplemental figure S3E). Furthermore, the generation of Tpex by RPE-IL12 appears to occur without the strong exhaustion signal54 observed with RPE-mIL2 or sham mice (figure 2I–K). Both cytokine factories led to a lower fraction of inhibitory Treg cells within the TME compared with RPE capsule control (online supplemental figure S3F–G). Moreover, Variable (V), Diversity (D), and Joining (J) analysis revealed that the T-cell repertoire present in RPE-mIL12 treated tumors was much more diverse than for RPE-mIL2 (figure 2L and online supplemental figure S3H,I). Notably, the T cells that underwent clonal expansion in RPE-mIL12 included both unique clonotypes and nearly all expanded clones present in RPE-mIL2 (figure 2M). The enhanced T-cell diversity seen with RPE-mIL12 may correlate with an improved antitumor response against both primary tumors and distal metastatic sites.9 55–59
Interactions between the CTLs recruited by cytokine factories and other immune cell types present at the TME will also influence the overall antitumor response. To understand these combinatorial interactions, we used NicheNet to assess signaling pathways through which other immune cell types communicate with CTLs and macrophages. These analyses revealed that RPE-mIL2 capsules primarily foster interactions between CTLs and dendritic cells (DCs), while RPE-mIL12 capsules promote a strong and inflammatory CTL-monocyte signaling axis (figure 2N and online supplemental figure S3J). Overall, RPE-mIL2 and RPE-mIL12 capsules promote different antitumor responses. This study suggests that the antitumor response induced by RPE-mIL2 capsules is driven by CTLs that are enriched for NK cells (figure 2O). In contrast, the antitumor response orchestrated by RPE-mIL12 capsules involves a cooperative interaction between T cells and inflammatory monocytes that creates an inflammatory TME with high T-cell diversity (figure 2P).
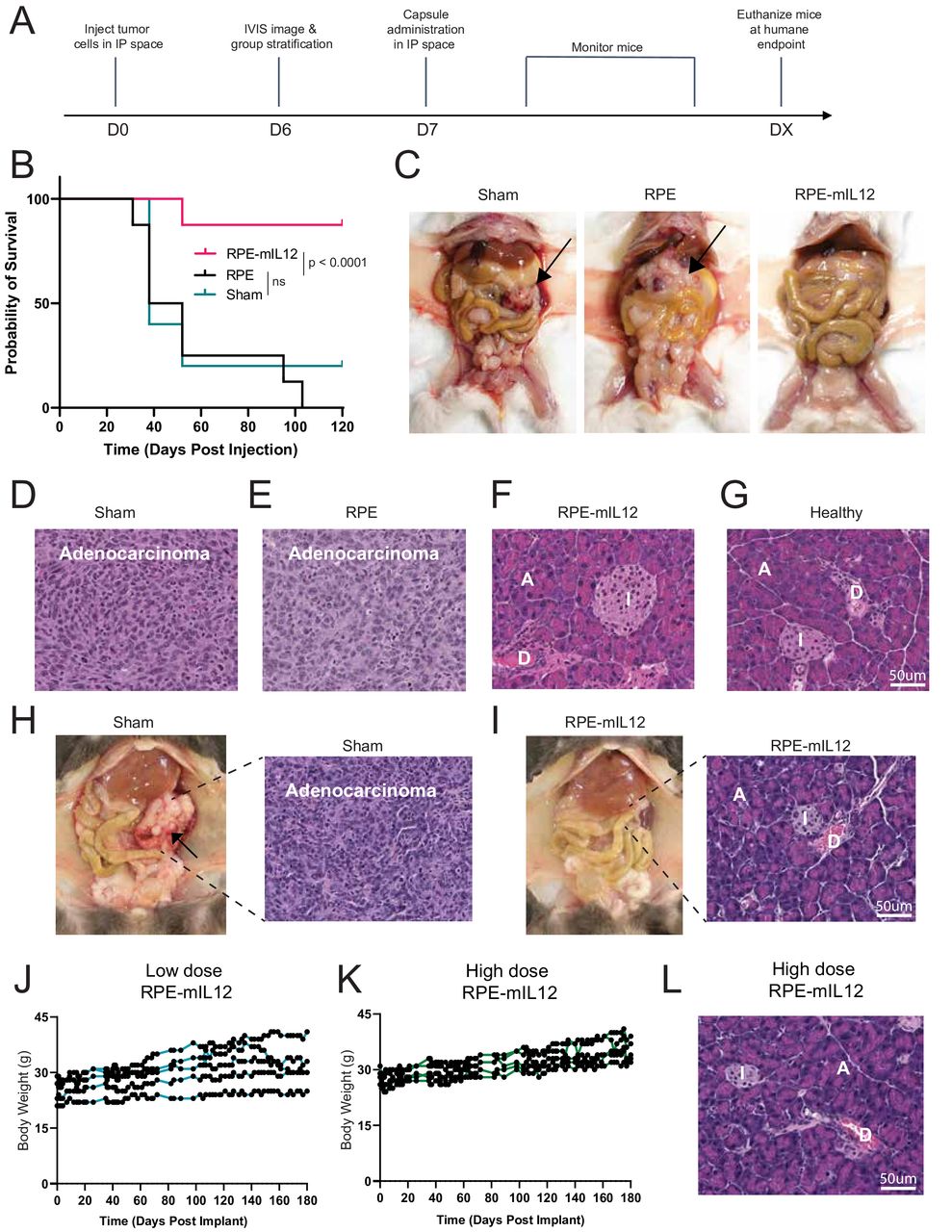
RPE-mIL12 treatment significantly extends the survival of mice with pancreatic tumors
To investigate the long-term effects of RPE-mIL12 treatment, we conducted a survival study. Following the experimental setup described above (figure 3A), we established PAN02 tumors in C57BL/6 mice (mixed gender) and treated them with RPE, RPE-mIL12, or sham. As animals reached humane endpoints, they were euthanized and macroscopically imaged (figure 3B). 120 days after treatment administration, all remaining animals were sacrificed and imaged. Representative images (figure 3C) highlight the ability of RPE-mIL12 treatment to eliminate pancreatic tumors in a safe and effective manner. Further, histological images of the pancreas at the time of sacrifice highlight the loss of anatomical features, such as islets, in the pancreatic tissue of animals treated with sham or RPE (figure 3D,E). Notably, the pancreas from animals treated with RPE-mIL12 showed identifiable islets, acinar cells, as well as ducts (figure 3F) and appeared similar to the pancreas collected from healthy naïve animals (figure 3G). 9/10 animals tolerated the RPE-mIL12 treatment for the duration of the experiment (online supplemental figure S4).

RPE-mIL12 treatment significantly extends the survival of mice with pancreatic tumors. (A) Experimental design for Pan02 intraperitoneal tumor model study. (B) Survival curves (n=8) depicted as percent survival over time in days after tumor inoculation. Comparison of survival curves was done using the log-rank, Mantel-Cox test. (C). Representative macroscopic images of the PAN02 pancreatic tumors inside the abdominal cavity of animals from each treatment group. Images were taken immediately after sacrifice. Black arrows indicate pancreatic tumors. (D–G) H&E staining of pancreas explanted from animals after sacrifice due to humane or scientific endpoints. Images shown are 20× magnification. The scale bar represents 100 um. A=acinar cells, D=duct, I=islet of Langerhans. (H) Representative macroscopic images of the KPC pancreatic tumors inside the abdominal cavity of animals from the sham or RPE-mIL12 treatment group. Images were taken immediately after sacrifice (8 weeks post-tumor inoculation). H&E staining of pancreas explanted from animals after sacrifice (to the right of each corresponding macroscopic necropsy image). (I) Body weight of animals bearing KPC pancreatic cancer (n=5) over time following intraperitoneal implantation of low-dose RPE-mIL12. (J) Body weight of animals bearing KPC pancreatic cancer (n=5) over time following intraperitoneal implantation of high-dose RPE-mIL12. (K) H&E staining of pancreas explanted from animals treated with high-dose RPE-mIL12 after sacrifice (180 days post-treatment). IP, intraperitoneal; IVIS, In Vitro Imaging System; ns, not significant; RPE, retinal pigmented epithelial cells.
To better understand the safety and breadth of the antitumor effects associated with RPE-mIL12 therapy, we conducted two long-term survival studies with a second pancreatic cancer cell line. In these studies, we established KPC pancreatic tumors in C57BL/6 mice (mixed gender) and waited 7 days for the tumors to develop and grow. Animals were then divided into two groups and treated with sham (untreated) or RPE-mIL12 and monitored for 56 days (study 1) or low or high-dose RPE-mIL12 and monitored for 180 days (study 2). Macroscopically, large vascularized pancreatic tumors were present in sham-treated animals at the time of sacrifice (figure 3H). Notably, the pancreas from animals treated with RPE-mIL12 showed identifiable islets, acinar cells, and ducts, while the pancreas collected from sham-treated animals did not have these characteristic cells (figure 3H,I). During these studies, we did not observe weight loss or any signs of toxicity, suggesting that RPE-mIL12 was well-tolerated long-term (figure 3J,K). Notably, at the conclusion of the study, we found no macroscopic or histological (figure 3L) evidence of tumors in animals from either dose group, suggesting that the therapy was highly effective at inducing the eradication of the tumor cells.
A robust and fully functional immune system is required for antitumor efficacy
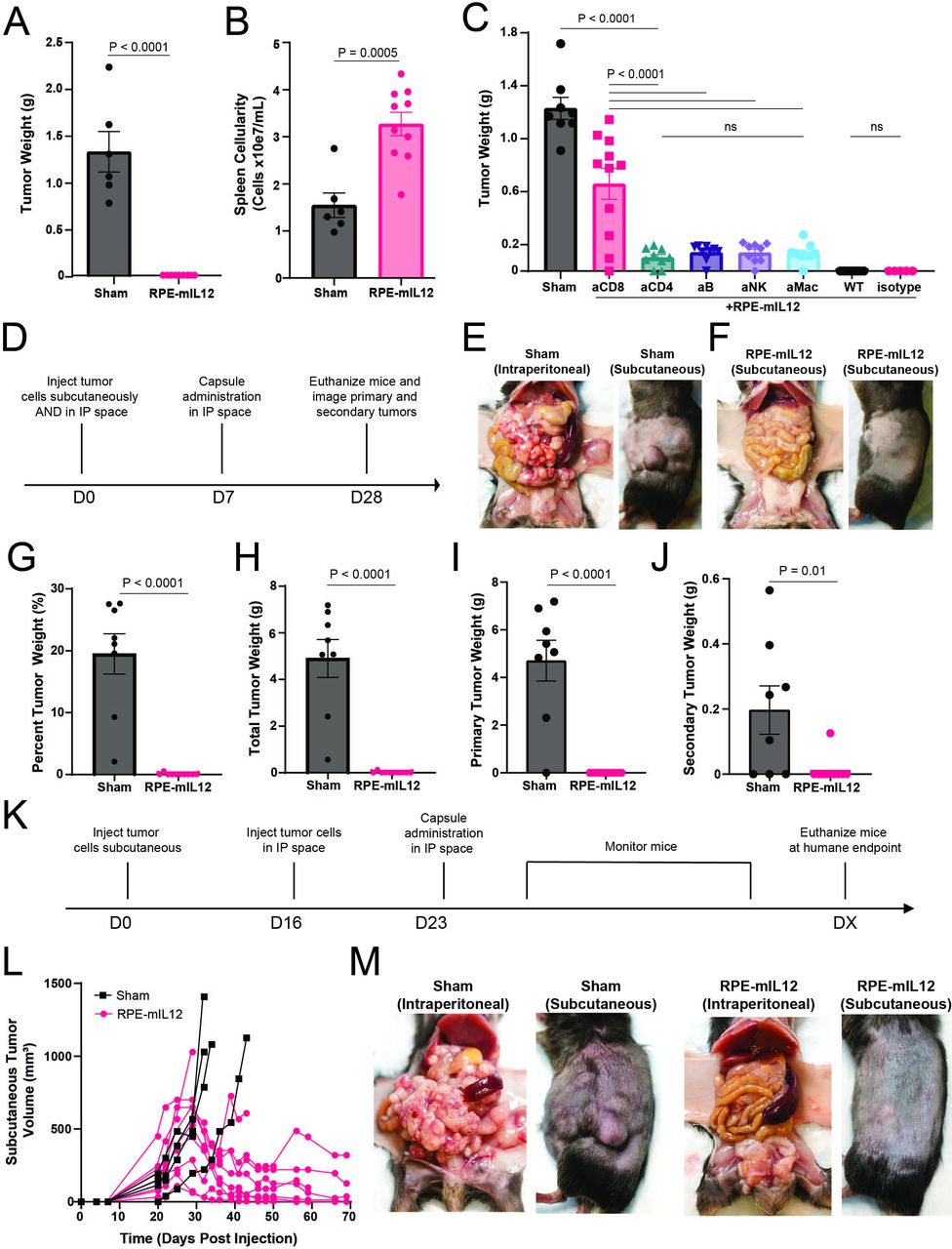
We next conducted an immune depletion study to investigate the components of the immune system required for the systemic antitumor response observed in the prior sections. To do this, we employed a mouse model of colorectal cancer as described previously.38 Unlike some of the other syngeneic models, MC38 colorectal cancer is highly immune infiltrated and thus amenable to the interrogation of individual immune subsets through depletion studies. First, we validated antitumor efficacy and bioactivity of RPE-mIL12 compared with sham mice in immunocompetent animals (figure 4A,B). Next, to selectively deplete immune cells, we split the mice into six groups and injected them with anti-CD4, anti-CD8, anti-CD20, anti-NK1.1, or anti-CSF1R. After two rounds of depletion injections, animals were treated with RPE-mIL12. After 1 week of treatment, the animals were sacrificed and the tumors were imaged, collected, and weighed. We found that animals lacking CD8+T cells had the largest tumors at the end of the study, suggesting that functional CD8+T cells are required for RPE-mIL12 treatment to be effective (figure 4C). Interestingly, we found that animals missing any of the five immune cells that we depleted were no longer able to fully eradicate the MC38 tumors (figure 4C). These data suggest that a robust and fully functional immune system is required for MC38 tumor eradication and further highlight the importance of our composite drug delivery system, which can activate innate and adaptive immune cells.

A fully functional immune system is required for antitumor efficacy and local RPE-mIL12 treatment can prevent tumor metastasis in mice. (A) Tumor weights (means±SEM) determined using an analytical balance. P values determined by t-test. (B) Spleen cellularity (means±SEM) determined by homogenizing spleens into a single cell suspension and counting using an automated cell counter. P values determined by t-test. (C) Tumor weights (means±SEM) determined using an analytical balance. (D) Experimental design for MC38 two-compartment tumor model study. (E–F) Representative macroscopic images of intraperitoneal (left) and subcutaneous tumors (right) from sham and RPE-mIL12 treated animals immediately after sacrifice. (G–J) Tumor weights (means±SEM) determined using an analytical balance and displayed as (G) a percentage of total body weight at the time of sacrifice, (H) total tumor weight, (I) primary (intraperitoneal) tumor weight, or (J) secondary (subcutaneous) tumor weight. (K) Experimental design for the second MC38 two-compartment tumor model study. (L) Subcutaneous tumor volume tracked over time for individual animals. (M) Representative macroscopic images of intraperitoneal (left) and subcutaneous tumors (right) from sham and RPE-mIL12 treated animals immediately after sacrifice. P values were calculated using one-way ANOVA with the Holm-Sidak method for multiple comparisons. Data are representative of two independent experiments. ANOVA, analysis of variance; IP, intraperitoneal; ns, not significant; RPE, retinal pigmented epithelial cells.
Local administration of RPE-mIL12 prevents the development of distal tumors
The data above suggest that RPE-mIL12 treatment induces a potent local antitumor effect on primary tumors restricted to one cavity. To determine whether this treatment could generate systemic immunity capable of eliminating more advanced metastases, we challenged the platform with a two-compartment colorectal cancer model. Briefly, we injected 1e6 MC38 cells into the IP cavity and 5e5 MC38 cells into the subcutaneous space on the right flank of C57BL/6 mice (mixed gender) and allowed the tumors to develop for 7 days (figure 4D). Animals were then randomly divided into two groups and treated with RPE-mIL12 or sham for 28 days. At the end of the study, animals were sacrificed, and the primary and secondary tumors were imaged, extracted, and weighed. Sham mice developed large tumors in both cavities (figure 4E and G–J) with 6/8 sham mice having nearly 20% of their final body weight being tumors. However, 9/9 RPE-mIL12 treated mice had no evidence of tumor in the IP cavity, and 8/9 mice did not develop subcutaneous tumors (figure 4F–J), suggesting that RPE-mIL12 treatment of local MC38 tumors protects from the development of secondary MC38 tumors.
Local administration of RPE-mIL12 causes reduction of pre-established distal tumors
To evaluate if local RPE-mIL12 treatment could reduce the size of pre-established distal tumors, we developed a second two-compartment colorectal tumor model. In this experiment, we injected 5e5 MC38 cells subcutaneously into the right flank of C57BL/6 mice and waited for subcutaneous tumors to establish and grow. 16 days after the subcutaneous injection, we injected animals with palpable subcutaneous tumors with 1e6 MC38 cells in the IP space and waited 1 week (figure 4K). Animals were then randomly divided into two groups and treated with RPE-mIL12 or sham and monitored three times per week until the length of the subcutaneous tissue surpassed 15 mm in any direction. The average survival time for the sham animals was 31.5 days (online supplemental figure S5A). The study concluded after 69 days, and 6/9 RPE-mIL12 treated animals remained alive with substantial subcutaneous tumor reduction (figure 4L). Notably, 5/6 RPE-mIL12 at the end of the study had tumors that were more than 3× smaller than prior to treatment, and 50% of the animals experienced complete subcutaneous tumor eradication. Further, sham animals also developed large, vascularized tumors in the IP cavity, whereas RPE-mIL12 treatment was also effective at IP tumor eradication (figure 4M). Finally, we did not observe any significant changes in animal weight (online supplemental figure S5B) suggesting that RPE-mIL12 treatment was well tolerated throughout the study. Collectively, these data suggest that local RPE-mIL12 treatment is well tolerated at therapeutically relevant doses and is able to induce a significant reduction of local and pre-established distal tumors when administered locally.
RPE-mIL12 delays the growth of metastatic melanoma tumors in the peritoneal cavity
To investigate the durability of RPE-mIL12 induced immune response within the context of an aggressive metastatic tumor, we used the melanoma metastasis mouse model described in figure 1. To quantify how long RPE-mIL12 therapy is able to delay the growth of this aggressive tumor, we conducted a survival experiment. As described above, we established the metastatic melanoma model and then treated animals with RPE-mIL12, RPE control, or sham and monitored them for signs of peritoneal tumor growth. As animals reached humane endpoints, they were sacrificed, and a survival curve was generated. While there was no measurable difference in survival time between the two control groups, RPE-mIL12 treatment significantly extended the animal survival time (greater than 2.3×) (online supplemental figure S6A). In addition, RPE-mI12-treated animals did not show signs of toxicity (eg, weight loss, (online supplemental figure S6B) throughout the experiment, which suggests that the RPE-mIL12 therapy was well tolerated. These data highlight the ability of RPE-mIL12 to significantly delay the growth of B16LN6 tumor cells in the peritoneal cavity and extend the survival time of animals, suggesting that this treatment has potential for clinical translation.
Recently, we showed that combining RPE-mIL2 therapy with aPD1 checkpoint therapy increased the antitumor efficacy of the cytokine factory platform in animals with malignant mesothelioma.40 Here, we hypothesized that combining RPE-mIL12 with aPD1 could also enhance the antitumor efficacy in our aggressive melanoma model. To test this hypothesis, we first asked whether animals would be able to tolerate this combination therapy and then whether the combination therapy would increase the therapeutic benefit of RPE-mIL12 alone. Briefly, we established the peritoneal metastasis model described above and treated animals with RPE-mIL12+aPD1, RPE+aPD1, or aPD1 injections. Notably, all animals (including the RPE-mIL12 treated animals) maintained similar body weights throughout the study, suggesting that the aPD1 injections were well tolerated (online supplemental figure S7A). Notably, however, there was a significant decrease in the weight (and appearance) of the tumors collected from RPE-mIL12+aPD1 treated animals at the conclusion of the study (online supplemental figure S7B), suggesting that the combination therapy was very effective against these aggressive tumors.
RPE-mIL12 in combination with PD1 checkpoint therapy reduces metastasis of melanoma to the lungs
The lungs represent a common site of distant metastasis for melanoma and many other solid tumors.60 61 To investigate whether RPE-mIL12 treatment could also treat metastases in the lung, we developed an additional two-compartment tumor model. In this model, we injected 1e5 B16LN6 cells into the peritoneal cavity of C57BL/6 mice (mixed gender). Four days later, we injected 2e5 B16LN6 cells into the tail vein of each animal and waited for tumor mets to grow in the lung (figure 5A). 7 days after the initial injection, animals were randomly divided into three groups and treated with RPE-mIL12, RPE-mIL12+aPD1, or sham. Animals were monitored for weight loss, abdominal cavity enlargement, and evidence of difficulty breathing and sacrificed when humane endpoints were met. At euthanasia, the IP tumors were collected and weighed, and the lungs were processed for histological analysis. Sham-treated animals had an average survival time of 20 days (figure 5B) and developed large tumor masses in the peritoneal cavity (figure 5C,D) as well as in the lungs (figure 5E). In contrast to the control group, all animals treated with RPE-mIL12 (n=10) or RPE-mIL12+aPD1 (n=10) remained alive and free from clinical symptoms for the entire study. In addition, we did not observe any significant deviations in baseline weight, suggesting that both therapies were well tolerated by the animals. The study was concluded after 35 days, and the animals were sacrificed as described above. Strikingly, RPE-mIL12 and RPE-mIL12+aPD1 treated animals had significantly smaller tumors in the peritoneal cavity (figure 5C,D) compared with sham controls. Interestingly, however, although RPE-mIL12 treatment was also very effective at reducing peritoneal tumor burden, RPE-mIL12+aPD1 treatment induced a more consistent antitumor response (figure 5C), suggesting that the combination therapy was better able to fully engage the host immune system in all animals. Taken together, these data highlight the profound antitumor effects of RPE-mIL12 and RPE-mIL12+aPD1 therapy and provide a rationale for the clinical evaluation of this platform in patients with aggressive cancer types.

RPE-mIL12 in combination with PD1 checkpoint therapy reduces metastasis of melanoma to the lungs. (A) Experimental design for two-compartment peritoneal metastatic melanoma model. (B) Percent survival over time (n=10/group). P value was determined by a comparison of survival curves by the log-rank (Mantel-Cox) test. (C) Primary (intraperitoneal) tumor weight (n=10/group), determined using an analytical balance. P value was determined using one-way ANOVA with the Holm-Sidak method for multiple comparisons. Data are representative of three independent experiments. (D) Representative macroscopic images of the melanoma tumors inside the abdominal cavity of animals from each treatment group. Images were taken immediately after sacrifice. (E) H&E staining of lungs explanted from animals after sacrifice due to humane or scientific endpoints. Images shown are 20× magnification. Scale bar represents 100 um. ANOVA, analysis of variance; aPD1, anti-programmed cell death protein-1; IP, intraperitoneal; RPE, retinal pigmented epithelial cells.
Local administration of RPE-hIL12 is well tolerated in a NHP
Finally, to assess the clinical translatability of this IL-12 delivery system, we developed RPE-hIL12 as described above and assayed for signs of toxicity in a NHP. Briefly, we evaluated body weight, temperature, platelet count, and kidney and liver function using complete blood count and blood chemistry analysis for more than 60 days in a cynomolgus macaque that was dosed with RPE-hIL12. On alginate implantation, there is transient immune activation that resolves over time, leading to stable pericapsular fibrotic overgrowth around capsules (figure 6A, left) and a predictable cessation of cytokine delivery. As seen in the previous studies, the hIL12 levels in the peritoneal cavity were significantly higher than the levels in the blood, demonstrating localization of the administered cytokine factories (figure 6A, middle-right). Notably, we found no significant changes in body weight (figure 6B, left) or deviations outside of the healthy ranges for body temperature (figure 6B, middle) or platelet count (figure 6B, right). To assess kidney function, we evaluated potassium, creatinine, and blood urea nitrogen levels over time and similarly found no significant deviations from healthy ranges (figure 6C). Last, to assess liver function, we evaluated aspartate aminotransferase, alanine aminotransferase (ALT), and gamma-glutamyl transferase levels over time. We found only one significant deviation (ALT level in week 6) from healthy ranges (figure 6D). This ALT spike was mitigated by the primate without clinical intervention and did not spike again for the remainder of the study, demonstrating that RPE-hIL12 was well tolerated. Taken together, the data in this work demonstrate the potential of RPE-IL12 as a versatile immunotherapeutic agent for a variety of solid tumors and support further clinical evaluation of this platform technology.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
RPE-hIL12 is well tolerated in the intraperitoneal space of a cynomolgus macaque. (A) Laparoscopic image of RPE-hIL12 capsules in the IP space of a cynomolgus macaque 61 days after administration. Black arrow points to the cytokine factory (left). Concentration of hIL-12 in the IP fluid (middle) and blood (right) over time. (B) Body weight, temp, and platelet count over time (n=1). (C) Creatinine, BUN, and potassium levels over time (n=1). (D) AST, ALT, and GGT levels over time (n=1). Red dashed lines indicate the healthy range for each biochemical parameter. The healthy range for non-human primate AST, ALT, GGT, BUN, potassium, and creatinine levels was taken from the study of Xie et al.66 ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; GGT, gamma-glutamyl transferase; IP, intraperitoneal; NHP, non-human primate.
Discussion/conclusion
Immunotherapy represents one of the greatest advances in oncology in decades, yet progress has stalled as many patients fail to generate systemic immune responses required to cure advanced-stage disease or develop intolerable off-target immune toxicities. Previously, we have developed a cell-based cytokine delivery platform for local cytokine production using alginate-encapsulated ARPE-19 cells engineered to secrete any cytokine of interest.38 We now demonstrate that using this technology to deliver the cytokine IL-12 leads to systemic immune activation and eradication of several aggressive peritoneal tumors, as well as models of subcutaneous and pulmonary metastases. RPE-IL12 monotherapy triggered durable responses in diverse mouse models, including metastatic melanoma, pancreatic cancer, and colorectal cancer, and was well tolerated. In addition to monotherapy, RPE-IL12 in combination with aPD1 therapy illustrated a high degree of synergy. The addition of checkpoint inhibitors to therapies capable of inducing systemic immunity has previously been shown to enhance responses against multiple tumors, suggesting the possibility that it broadens the antigens recognized by T-cell responses.6 Similarly, the addition of aPD1 to the RPE-IL12 therapy may further broaden the T-cell responses, particularly against metastatic disease. Together, our investigation of RPE-IL12 monotherapy and combination therapy supports its safety and versatility in the treatment of advanced malignancies.
We hypothesize that the success of RPE-IL12 in treating challenging cancer models is due to the robust immune response elicited. Compared with RPE-IL2 therapy, RPE-IL12 therapy leads to the generation of a more diverse T-cell repertoire evident through the greatly increased number of unique T-cell clonotypes generated. For these studies, we elected to use highly aggressive tumor models, many of which are unresponsive to existing immune checkpoint inhibitors due to a milieu of immunosuppressive features. Furthermore, to better approximate the clinical scenario, we employed metastatic implementations of these preclinical models. The ability of RPE-IL12’s ability to sustain broad T-cell responses minimizes the probability of immune evasion, making it well suited for treatment of these advanced cancer types. Furthermore, our results are the first to suggest that IL-12 is capable of inducing Tpex at enhanced levels compared with other cytokine therapies. Induction of Tpex populations has been identified as a critical mediator of effective immune responses to checkpoint inhibitors, where maintenance of a sustained Tpex population within tumor-draining LNs, and additional priming by classical DC type I populations within the TME, distinguishes robust immune responses from those that ultimately falter.21 31–35 62 The upregulation of CXCR3 ligands CXCL9/10, DC activation markers (eg, CD80), and interferon-stimulated genes, rather than a simple expression of effector cytotoxic T-cell molecules (eg, granzymes) is suggestive of a robust sustainable immune response that is unlikely to become dysfunctional. Perhaps, the high degree of success in our mouse models is a consequence of such Tpex induction or recruitment, in addition to the diverse T-cell repertoire generated. While IL-12 has proven to be a potent cytokine for eliciting strong antitumor responses, translation of IL-12-based therapy is challenging due to narrow therapeutic windows and broad toxicity profiles. Our engineered cytokine factory approach overcomes these limitations by generating locally high concentrations of IL-12 adjacent to the tumor while maintaining nearly undetectable levels in the blood. In doing so, we are able to deliver a therapy capable of inducing potent and durable antitumor immunity that eradicates metastases while maintaining a safety profile that is likely to be highly tolerated in patients.
IL-12 has yielded profound antitumor effects in a variety of mouse models, but its applications in humans have been limited by its significant toxicity profile.63 64 This has motivated a shift towards the development of targeted or local delivery of IL-12. However, several IL-12 delivery technologies including engineered IL-12 proteins,65–73 gene therapies 74–78 and cell therapies79–81 are in development intended to maximize tumor-adjacent IL-12 concentrations and limit systemic exposure. The first published study on IL-12-producing cell therapy was released in 1996, where Zitvogel and colleagues isolated dendritic cells from the bone marrow of mice and engineered them to secrete murine IL-12. These cells were then injected intravenously to treat mice inoculated with MCA205 tumor cells. While mice receiving treatment were never completely cured, disease stabilization was observed.82 Since this study has been published, multiple clinical trials have been initiated investigating the treatment of patients via injection of autologous cells, including tumor-infiltrating lymphocytes, T cells, and fibroblasts, engineered to secrete IL-12. Transient antitumor responses were observed, but these technologies were often limited by severe toxicities, variable IL-12 pharmacokinetics following treatment, or rapid clearance of the injected cells over time.79–81 The technology described in this manuscript provides several advantages compared with the IL-12 cell therapies tested in the clinic to date. By encapsulating IL-12 producing ARPE-19 cells prior to implantation, we achieve greater control over the dose of IL-12 administered and introduce a barrier to the foreign body response, greatly improving the duration of treatment. Furthermore, engineering allogeneic ARPE-19 cells rather than patient-derived cells may improve the availability of treatment by reducing the time, expertize, and costs required to isolate, modify, and reinfuse the engineered cells. Together, these aspects of RPE-IL12 therapy may overcome the limitations of current IL-12 cell therapies and allow for improved outcomes in humans.
Our results here demonstrate that constitutive delivery of IL-12 with our technology yields promising preclinical results; however, this cytokine delivery platform can be applied to a variety of different contexts. We have currently demonstrated that RPE therapy can effectively deliver IL-2 and IL-12 therapy in a safe manner, and this platform can be expanded to deliver a wide range of other biologics as well. Furthermore, cell engineering can be leveraged to develop regulated versions of this treatment. For example, small molecule control of protein production with the Food and Drug Administration (FDA)-approved small molecule trimethoprim has been demonstrated in vivo.83 Our technology is amenable to using this or similar methods of small molecule activation to allow for conditional dosing control. In addition to small molecule control, ARPE-19 cells can also be engineered to contain regulatory circuits for feedback control. Production of a therapeutic can be linked to a biomarker of interest such that dosing is modified based on the treatment mechanism of action. In the context of IL-12 delivery, a potential application of regulated RPE-IL12 therapy would be the regulation of IL-12 production based on the biomarker IFN-γ. By downregulating IL-12 production in the presence of large concentrations of IFN-γ, severe toxicities could potentially be avoided by lowering doses once the immune system has been sufficiently activated. Overall, we believe our technology can potentially allow for advanced immunotherapy delivery through the application of these cell engineering concepts.
Here, we illustrated that RPE-IL12 therapy is highly tolerable and successfully eradicates both primary peritoneal and metastatic tumors in multiple advanced mouse cancer models. We believe that the efficacy of treatment is largely due to RPE-IL12’s ability to induce Tpex cells, a feature that may be promising in preventing T-cell exhaustion following IL-12 treatment and generating a systemic immune response. To date, no IL-12-based therapies have advanced past phase II clinical trials, which largely seems to be due to the complex immune response generated by IL-12.75 84 85 Our results further characterize IL-12’s effects on the immune system, which will potentially allow for the improved design of IL-12-based therapies. In the context of peritoneal cancers, due to the high tolerability, efficacy, and robust immune response created, we believe our results provide strong rationale for human testing of RPE-IL12 and that this platform has the potential to become the first FDA-approved IL-12-based immunotherapy for patients with peritoneal tumors.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors acknowledge funding support for this work through an Avenge Bio Sponsored Research Award (SRA) to Rice University (to OV), Cancer Prevention Research Institute of Texas grant RR160047 (to OV), National Institute of Health R01 grant R01CA272769 (to HCH) and DP2 grant DP2 AI177915 (to NR-F), and the Advanced Research Projects Agency for Health (ARPA-H) under Award Number AY1AX000003 (to OV). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Advanced Research Projects Agency for Health or the National Institute of Health.
References
Footnotes
X @hodgeshc, @retickerflynn
Contributors ANa and OV designed the studies. ANa, DM, JD, BC, BK, FH, CC, ANg, AH, ZW, PDR, SG, IJ, DI, NZ, conducted the experiments, analyzed data, carried out statistical analyses, and prepared displays communicating datasets. WP, OAI, JO, HCH, NR-F, and OV provided advice and technical support throughout the studies. OV supervised the study. ANa wrote the paper with assistance from JD, WP, OAI, JO, HCH, NR-F, and OV. All authors discussed the results and the preparation of the paper. OV is the guarantor
Funding This study was funded by Cancer Prevention Research Institute of Texas (RR160047), Advanced Research Projects Agency for Health (ARPA-H) (AY1AX000003), Center for Cancer Research (R01CA272769), Common Fund (AI177915), SRA.
Competing interests ANa and OV declare interests via patents filed by Rice University on the technology described in this manuscript. ANa, JO, NR-F, and OV hold equity in Avenge Bio. The authors declare that they have no other competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.