Article Text

Abstract
Background Invariant natural killer T (iNKT) cells and CD8+ T cells are key in the immune response against multiple myeloma (MM), a largely incurable blood cancer. Immunization is a promising strategy to activate these T cell populations. To our knowledge, immunization with messenger RNA (mRNA) and the iNKT agonist, α-galactosylceramide (αGC), has not been studied in MM, as knowledge on clinically relevant antigens in preclinical MM models is lacking.
Methods Microarray data and immunopeptidomics (imPep) were used to identify candidate antigens for immunization in 5TMM models. Galsomes, lipid nanoparticles containing antigen mRNA and αGC were used to immunize 5T33MM-bearing mice. This treatment was combined with a CD40 agonist. Tumor burden and activation of iNKT cells and CD8+ T cells were studied using M-protein electrophoresis, flow cytometry and ELISA.
Results RNA transcripts revealed survivin as a candidate antigen. Prime-boost Galsomes therapy targeting survivin significantly reduced M-protein levels despite low survivin-specific T cell responses. Further analysis showed potential T cell fratricide. ImPep revealed HSP60, Idiotype, PICALM and EF1A1 as candidate antigens. Prime-boost therapy with Galsomes targeting these antigens reduced MM growth significantly when combined with a CD40 agonist, coinciding with significantly improved antigen presentation, costimulation and cytotoxicity of iNKT cells and CD8+ T cells.
Conclusion These findings highlight the potential of Galsomes, an mRNA vaccine designed to activate CD8+ T cells and iNKT cells, for MM therapy, and emphasize the importance of combinatorial approaches, addressing immune anergy for effective MM immunotherapies.
- Multiple Myeloma
- Vaccine
- Major histocompatibility complex - MHC
- Nanoparticle
- Immunotherapy
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request. The mass spectrometry proteomics data, with identifier PXD060156, have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://proteomecentral.proteomexchange.org/PXD060156). The necessary peptide data is included in supplementary tables. Other data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Clinical trials that evaluate therapeutic vaccines to treat multiple myeloma (MM) are ongoing. These are based on proteins, peptides, engineered dendritic cells (DCs) or MM cells, focusing on CD8+ T cell activation. Their combination with immunomodulatory drugs (eg, lenalidomide, immune checkpoint inhibitors) is studied to improve therapy outcome.
WHAT THIS STUDY ADDS
Therapeutic messenger RNA (mRNA) vaccines have not yet been studied in MM. For this study, we identified vaccine antigens using RNA transcript levels and the cutting-edge immunopeptidomics (imPep) technology. The clinical relevance of these antigens was demonstrated using the patient cohort Total Therapy 2. We employ Galsomes—lipid nanoparticles containing antigen mRNA and α-galactosylceramide—to activate CD8+ T cells and invariant natural killer T (iNKT) cells. The preclinical data advise against survivin as an antigen, but show the potential of the combination therapy of a CD40 agonist and imPep-based mRNA vaccine as a personalized treatment.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This preclinical study raises caution regarding the use of survivin as a vaccine antigen due to the observed T cell death. However, it highlights the potential of therapeutic vaccines that broaden the scope from CD8+ T cells to iNKT cells as well as the potential of imPep and mRNA to develop such a vaccine, especially when combined with DC stimulators such as CD40 agonists. This is also relevant in other mRNA vaccine settings (eg, COVID-19 vaccines) in patients with MM, where impaired cellular responses are noted.
Introduction
Multiple myeloma (MM) is a blood cancer characterized by the clonal expansion of malignant plasma cells that produce immunoglobulins (M-protein, paraprotein or idiotype) in the bone marrow (BM).1 Treatment options for patients with MM range from high-dose chemotherapy and autologous stem cell transplantation to proteasome inhibitors and more recently immunotherapy strategies, such as adoptive transfer of MM-specific monoclonal antibodies (mAbs), bispecific T cell engagers or chimeric antigen receptor T cells.2–4 Due to advances in the treatment of patients with MM, the median overall survival in MM is reaching beyond 10 years. Still, MM remains largely incurable because many patients with MM experience relapse as a result of drug-resistant MM cells.1
T cells, in particular MM-specific CD8+ cytotoxic T lymphocytes and invariant natural killer T (iNKT) cells, hold the promise of controlling MM cells.5–8 Therefore, activating these T cell populations through therapeutic vaccination constitutes an elegant approach to treat patients with MM. We previously described Galsomes, lipid nanoparticles (LNPs) that contain tumor antigen messenger RNA (mRNA) and α-galactosylceramide (αGC).9–11 Galsomes transfect antigen-presenting cells (APCs) on in situ delivery, enabling presentation of the glycolipid αGC in CD1d for presentation to iNKT cells as well as presentation of the mRNA-encoded tumor antigen in major histocompatibility class I molecules (MHC-I) to CD8+ T cells. Vaccination with Galsomes has been shown effective in mouse models of ovalbumin-positive melanoma and T cell lymphoma. Still, their evaluation in mouse models of MM, a necessity to move towards clinical evaluation, has been hampered due to a lack of well-defined target tumor antigens.
In this study, we used the mouse 5T33MM model, a fully immunocompetent mouse model that closely resembles human MM, to evaluate Galsome therapy in MM. We first analyzed the antigenic landscape of the 5T33MM model on the transcriptome level, selecting survivin as a target antigen, given its proven ability to elicit T cell responses and clinical benefit in a number of patients with MM.12 13 We observed a statistically significant decrease in M-protein serum levels following prime-boost Galsome therapy. Following boost vaccination, we observed lower iNKT and survivin-specific CD8+ T cell numbers and an overall lower potency of CD8+ T cells to produce interferon-gamma (IFN-γ), suggesting survivin-specific fratricide or activation-induced cell death (AICD),14 15 confirmed by combining survivin and ovalbumin mRNA in prime-boost Galsome therapy. Therefore, we subsequently performed immunopeptidomics (imPep) to analyze the antigen landscape on the MHC ligand level using 5TGM1 and 5T33vt, both cell lines derived from the 5T33MM model. Using selected clinically relevant MM antigens for Galsome vaccination, a statistically significant reduction in M-protein levels correlating with increased cytotoxic iNKT and CD8+ T cells, positive for programmed death-1 (PD-1) and CD107a was observed when combining Galsome vaccination with mAbs activating CD40 on APCs. These data underscore the potential of MM antigen mRNA containing Galsomes as therapy in MM, especially when combined with dendritic cell (DC) stimulators such as CD40 agonists. This is also relevant in other mRNA vaccine settings (eg, COVID-19 vaccines) in patients with MM, where impaired cellular responses are noted.
Material and methods
5T33MM and 5TGM1 models
The 5T33MM (in vivo) MM model, developed by the Radl group,16 is a syngeneic, immunocompetent MM model which was propagated in young C57BL/KaLwRij mice (Envigo) by intravenous transfer of 1.0×105 5T33 MM containing BM cells of diseased mice. This model is derived from spontaneously arising MM in aging C57BL/KaLwRij mice.
For the 5TGM1 model, a total of 1.0×106 5TGM1 cells were intravenously injected. 5TGM1 and 5T33MMvt (in vitro) cell lines were cultured at 37°C and 5% CO2 in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma-Aldrich) supplemented with 100 U/mL penicillin, 100 µg/mL streptomycin, 1 mM sodium pyruvate, 2 mM L-Glutamine (Sigma-Aldrich) and 10% fetal bovine serum (PAN Biotech), referred to as RPMI 1640+. Mice were randomly assigned to cages and housed in the animal facility of VUB Campus Jette under standard conditions (temperature, humidity, and light/dark cycle). Following injection with 5T33MM or 5TGM1 cells, cages were randomly allocated to treatment groups. Sample sizes were determined using G*Power based on tumor effect sizes (primary outcomes) and were reviewed by the ethical committee’s statistics team. Sample sizes for each experiment are provided in the figure legends. Potential confounders were not controlled in this study. Additional details on treatment schemes and in vitro testing can be found in the results and supplemental methods.
Gene expression analysis
The 5T33MM microarray data is accessible via the ArrayExpress database (E-MTAB-3178).17 Gene expression profiling data of Total Therapy 2 (TT2), a microarray-based gene expression dataset of 345 newly diagnosed patients with MM (GSE2658) and 22 healthy donors, 44 monoclonal gammopathy of unknown significance (MGUS) and 12 cases of smoldering myeloma (SMM) (GSE5900) from the University of Arkansas for Medical Sciences were consulted and analyzed via GenomicScape (http://www.genomicscape.com).18–20 Data were normalized with the MAS5 algorithm.
Immunopeptidomics
Isolation, purification, Tandem Mass Tag (TMT) labeling, prefractionation, Liquid Chromatography (LC)–mass spectrometry (MS)/MS and immunopeptide data analysis were performed as described using 1.8×1e8 5T33MMvt and 4×1e8 5TGM1 cells as starting material.21 Detailed information is included in online supplemental material and methods.22–25
Supplemental material
Galsome formulation
Galsome production started with preparing in vitro transcribed mRNA. The plasmids pGEM-tNGFR and moIi80tOVA encoding truncated nerve growth factor receptor and truncated ovalbumin fused to the first 80 amino acids of the mouse invariant chain were described.26 The plasmid pLMCT was used to clone gBlocks for murine BIRC5 (survivin), EF1A1, PICALM, HSP60 and IgK (IDT) as described.27 In vitro transcription of mRNA was done as earlier described27 using 5-methylcytidine and N1-methyl-pseudouridine nucleotides (TriLink). This mRNA was formulated in Galsomes, LNPs containing the ionizable lipid C12-200, cholesterol, DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), DMG-PEG 2000 (1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000) and αGC at a component molar ratio of 50/10/38.5/1.5/0.02.9 The detailed information is included in online supplemental material and methods.
Flow cytometry
Cells were incubated for 30 min in phosphate-buffered saline (PBS) with Fc blocking CD16/32 antibodies (CD16/32, BioLegend) and eF506 viability dye (eBioscience) in PBS. Cells were washed and antibody labeled in PBS with 1% bovine serum albumin (Merck Millipore) and 0.02% sodium azide (Sigma-Aldrich). Antibodies, dextramers are available in online supplemental material and methods. Cell acquisition was performed on the BD LSRFortessa or BD Symphony A1 flow cytometers. Data collection and analysis were done using the FlowJo software V.10.7.1 (Tristar). Used gating strategy is included as online supplemental figure 1.
Supplemental material
Disease load
Serum M-protein levels (g/L) were determined by serum electrophoresis, performed at the Clinical Chemistry Department of the UZ Brussel (Belgium).24
Statistical analysis
Statistical analysis was performed using GraphPad Prism V.10.0 software. The statistical test used for a dataset is indicated in the figure legends. Statistical significance is indicated by the p value. Figures were generated using BioRender and GraphPad Prism V.10.0 software.
Results
Survivin is a potential target in 5T33MM cells due to its high biological and clinical relevance
To identify suitable candidate antigens to test therapeutic mRNA vaccination in MM, we reused available microarray data from the 5T33MM mouse model (E-MTAB-3178).17 We evaluated gene expression of Birc5 (survivin), Ms4a1 (CD20), Dkk1 (DKK-1), Tnfrsf17 (BCMA), and Slamf7 (CS1) in 5T33MM cells (figure 1A), as potential target antigens in the 5T33MM mouse model, since antigen-specific T cells targeting peptides derived from these proteins have been detected in patients with MM.28–31 Among the selected antigens, survivin (Birc5) emerged as the highest expressed, as indicated by the highest Log2 expression value. Because of its high expression in 5T33MM cells we further validated its relevance by analyzing gene expression data from a cohort of patients with MM. Gene expression data from the TT2 cohort was used to compare BIRC5 expression in plasma cells during disease progression (GSE2658, GSE5900).19 Plasma cells from healthy individuals and patients with MGUS or SMM showed low expression, while heterogeneous BIRC5 mRNA expression was observed in patients with active MM disease (figure 1B). Furthermore, high expression of BIRC5 mRNA is correlated with significantly lower overall survival of patients with MM (figure 1C) (GSE2658).19

Survivin is a target antigen in 5T33MM cells with clinical relevance. (A) Log2 values of transcript levels found in four 5T33MM-bearing mice. (B) Scatter plot showing BIRC5 (survivin) expression in BM plasma cells of healthy donors (BMPC) and donors diagnosed with MGUS, smoldering MM (SMM) or MM in the TT2 cohort. Data are log transformed, MAS5-normalized Affymetrix signals with each data point representing one donor (n=12–345). Statistical significance was determined by one-way ANOVA (Tukey post hoc); two-sided p values are reported. (C) Kaplan-Meier curves for patients with MM with low (blue, n=279) or high (red, n=66) expression of BIRC5 (survivin, probe 20,095) in the TT2 cohort. MaxStat analysis was used to calculate the optimal separation of patients based on a cut-off value. The log-rank (Mantel-Cox) test was used to analyze survival curves (n=345). ANOVA, analysis of variance; BM, bone marrow; MGUS, monoclonal gammopathy of unknown significance; MM, multiple myeloma; TT2, Total Therapy 2.
Intravenous vaccination with Galsomes containing survivin mRNA impacts MM progression despite absence of survivin-specific CD8+ T cells
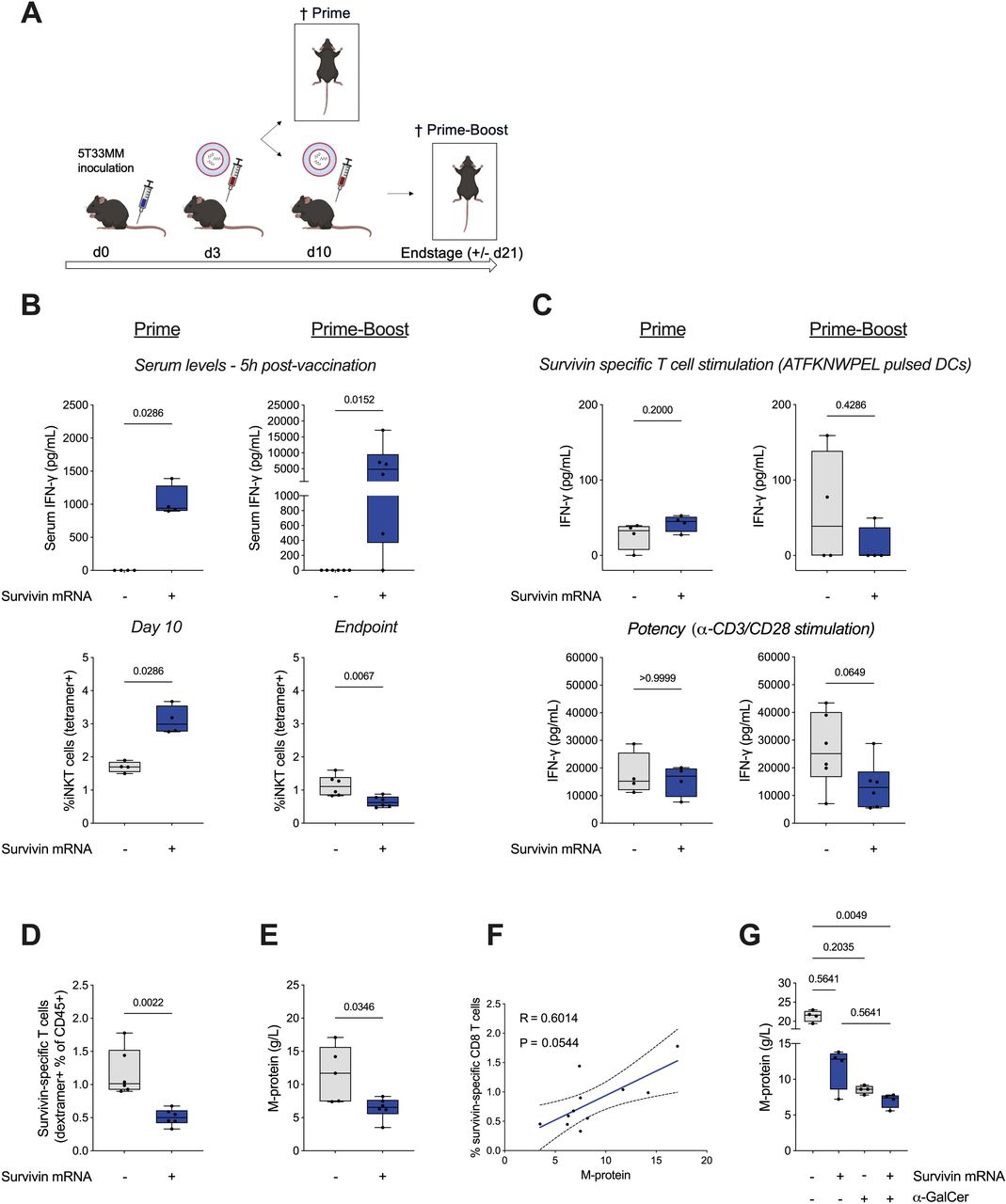
To address the potential of Galsome vaccination in the 5T33MM model, we vaccinated 5T33MM bearing mice intravenously with Galsomes containing 2.5 µg survivin mRNA on day 3 (prime) or on day 3 and 10 (prime-boost) post tumor cell inoculation (figure 2A). As a control, a separate cohort of mice was injected intravenously with PBS. We quantified IFN-γ by ELISA in serum collected 5 hours after vaccine delivery, showing high IFN-γ serum levels both following prime and boost vaccination, suggestive of strong iNKT cell activation (figure 2B, upper panel). Flow cytometry analysis of splenocytes, isolated 7 days after prime vaccination or 11 days after prime-boost vaccination, showed a significant increase in iNKT cells after prime vaccination, while a significant decrease of iNKT cells was observed following boost vaccination (figure 2B, lower panel). To study the presence of survivin-specific CD8+ T cells, we restimulated CD8+ splenocytes, isolated 7 days after prime vaccination or 11 days after prime-boost vaccination, with DCs pulsed with survivin peptide (ATFKNWPEL) (figure 2C). Moreover, we stimulated the CD8+ splenocytes non-specifically with anti-CD3/anti-CD28 antibody coated beads to study the potency of the CD8+ T cells to produce IFN-γ. Following prime vaccination, we observed low levels of IFN-γ when CD8+ splenocytes from PBS or Galsome vaccinated mice were stimulated by survivin presenting DCs with a trend in increased IFN-γ production by CD8+ splenocytes from Galsome vaccinated mice (figure 2C, upper panel). No difference in IFN-γ production was observed for CD8+ splenocytes from Galsome and PBS-treated mice when non-specifically (anti-CD3/anti-CD28) stimulated (figure 2C, lower panel). Contrary to expectations, production of IFN-γ by CD8+ splenocytes from Galsome vaccinated mice was not increased after boost vaccination, instead we observed little IFN-γ production following survivin-specific or non-specific restimulation. Flow cytometry showed fewer survivin-specific CD8+ T cells in mice vaccinated with a prime-boost Galsome regimen compared with mice receiving PBS injections (figure 2D). Still, we observed that M-protein levels, a measure of tumor burden, were statistically significantly reduced in Galsome prime-boost compared with PBS-treated mice (figure 2E). Interestingly, the reduction in survivin-specific T cells seems to correlate (p=0.0544) with a decrease in tumor load (figure 2F).

Vaccination with Galsomes containing survivin mRNA impacts MM progression. (A) Schematic representation of the experimental set-up. C57BL/KalwRij mice were inoculated with 5T33MM containing BM cells on day 0. An intravenous injection of Galsomes (+, blue) or PBS (−, gray) was performed on day 3 (prime). On day 10, four mice were sacrificed, while six mice received a second intravenous injection of Galsomes or PBS (prime-boost). Prime-boost vaccinated mice were sacrificed on day 21 when the first mouse in the experiment reached humane endpoints. (B) Box plot graphs showing serum IFN-γ levels as determined 5 hours after prime or boost vaccination (top) or showing the percentage of iNKT cells within splenocytes isolated on day 10 (prime) or 21 (prime-boost) (bottom). (C) Box plot graphs showing IFN-γ production by CD8+ splenocytes as determined 24 hours after survivin-specific (top) or non-specific (anti-CD3/CD28) (bottom) stimulation after prime (left) or boost (right) vaccination. (D) Box plot showing the percentage of survivin-specific CD8+ T cells within splenocytes isolated on day 21 (prime-boost). (E) Box plot showing M-protein levels in serum that was collected on day 21 (prime-boost). (F) Correlation between survivin-specific T cells and disease load (M-protein). Spearman’s rank correlation coefficient (R) and the 95% CI (dashed lines) are shown. In B–E, the gray and blue box plots show the results of mice treated with PBS and Galsomes respectively (n=4 for prime, n=6 for prime-boost). (G) Box plot showing M-protein levels in serum that was collected on day 21 (prime-boost) of mice vaccinated with PBS, LNPs encapsulating survivin mRNA, Galsomes with moIi80tOVA mRNA and Galsomes with survivin mRNA (n=4/group). The horizontal line shows the mean, while each dot represents one data point or mouse. Statistical significance was determined using the Mann-Whitney U test and for (G) the Kruskal-Wallis test. Two-sided p values are shown. α-GalCer, α-galactosylceramide; BM, bone marrow; DC, dendritic cell; IFN-γ, interferon-gamma; iNKT, invariant natural killer T cell; LNPs, lipid nanoparticles; MM, multiple myeloma; mRNA, messenger RNA; PBS, phosphate-buffered saline.
To further investigate the roles of iNKT cells and survivin-specific CD8+ T cells in the antitumor response, we vaccinated mice in a prime-boost manner intravenously with either PBS (control), Galsomes with αGC and survivin mRNA (stimulating both survivin-specific CD8+ T cells and iNKT cells) or LNPs with survivin mRNA but without αGC (stimulating survivin-specific CD8+ T cells). To activate iNKT cells without engaging tumor-specific CD8+ T cells, we treated mice with LNPs containing moIi80tOVA mRNA with αGC. The inclusion of moIi80tOVA mRNA allowed iNKT cells to interact with activated CD8+ T cells without CD8+ T cells contributing to tumor cell rejection, as OVA is not an antigen in the 5T33MM model. Mice were sacrificed at end stage, and we observed a reduction in M-protein levels (figure 2G) across all three treatment groups, with the most substantial reduction seen in mice treated with Galsomes containing survivin mRNA (mean: 7.05 g/L, in comparison to PBS: 21.4 g/L, LNPs with survivin mRNA 11.65 and Galsomes with moIi80tOVA 8.55 g/L), showing that both CD8+ T cells and iNKT cells contribute to the antitumor effect despite their numeric loss.
The Galsome vaccination route and use of survivin mRNA impacts CD8+ T cells
The loss of iNKT and CD8+ T cells after prime-boost Galsome vaccination in the 5T33MM model prompted us to address whether this is due to disease progression, anergy or the antigen survivin. We repeated prime-boost Galsome vaccination in healthy mice, excluding any impact of the disease. Both intravenous and intramuscular injection routes were evaluated, as repeated local versus intravenous administrations of αGC were shown by others to reduce iNKT cell anergy.32–34 We also compared Galsomes with 2.5 µg moIi80tOVA mRNA supplemented with 2.5 µg tNGFR or survivin mRNA to address whether stimulation of survivin-specific T cells could lead to T-cell death.14 We chose to study ovalbumin-specific T cells, as ovalbumin is a strong immunogen. Healthy mice were vaccinated on day 0 (prime) or day 0 and 14 (prime-boost) (figure 3A). We analyzed iNKT and CD8+ T cell numbers 7 days after the last treatment. Results were normalized to the PBS group to facilitate comparison of the prime and prime-boost outcome following intravenous or intramuscular Galsome vaccination. We observed that iNKT cell numbers were not significantly impacted by the inclusion of survivin mRNA into the Galsomes (figure 3B and C). However, following prime-boost vaccination we observed a significant decrease in iNKT cell numbers in mice vaccinated intravenously (prime vs prime-boost, figure 3B), which was not observed after repeated intramuscular vaccination (figure 3C). Ovalbumin-specific CD8+ T cell levels were not affected by the inclusion of survivin mRNA after one (prime) intravenous or intramuscular Galsome vaccination (figure 3D and E). However, the initial magnitude of specific T cells was greater in intravenously vaccinated mice. A clear prime-boost benefit was observed following intramuscular boost vaccination with Galsomes containing moIi80tOVA and tNGFR or survivin mRNA, achieving a magnitude of ovalbumin-specific CD8+ T cells comparable to that seen after intravenous prime vaccination (figure 3D and E). Notably, following intramuscular boost vaccination, Ovalbumin-specific CD8+ T cell levels were significantly lower in mice treated with Galsomes containing survivin mRNA compared with Galsomes containing moIi80tOVA and tNGFR mRNA (figure 3E). We further studied the capacity of CD8+ T cells to produce IFN-γ on stimulation with the ovalbumin immunodominant peptide (SIINFEKL) pulsed DCs. We showed a strong significant decrease in IFN-γ production following intravenous boost vaccination with Galsomes containing moIi80tOVA and tNGFR or survivin mRNA (figure 3F). In contrast, CD8+ T cells isolated after intramuscular boost vaccination demonstrated significantly higher IFN-γ production compared with prime intramuscular Galsome vaccination (figure 3G). These data confirm that intravenous Galsome vaccination induces iNKT loss and CD8+ T cell anergy. Furthermore, the inclusion of survivin mRNA in Galsomes appears to negatively impact CD8+ T cell numbers, highlighting the need to explore alternative antigen candidates in conjunction with intramuscular Galsome vaccination.

The Galsome vaccination route and use of survivin mRNA impacts CD8+ T cell responses. (A) Schematic representation of the experimental set-up. C57BL/6 mice were vaccinated intravenously or intramuscularly with Galsomes containing moIi80tOVA and tNGFR mRNA (OVA, light blue) or with moIi80tOVA and survivin mRNA (Survivin, dark blue). On day 7, five mice were sacrificed for analysis (prime), while the five remaining mice received a second vaccine (prime-boost). The prime-boost vaccinated mice were sacrificed on day 14. Results were normalized to the corresponding PBS group. (B–C) Box plot graphs showing the relative number of iNKT cells within splenocytes isolated on day 7 (prime) or 14 (prime-boost) from mice receiving an intravenous (B) or intramuscular (C) injection with Galsomes containing the indicated mRNA. (D–E) Box plot graphs showing the relative number of ovalbumin-specific CD8+ T cells within splenocytes isolated on day 7 (prime) or 14 (prime-boost) from mice receiving an intravenous (D) or intramuscular (E) injection with Galsomes containing the indicated mRNA. (F–G) Box plot graphs showing IFN-γ production by CD8+ splenocytes as determined 24 hours following ovalbumin-specific restimulation after intravenous (F) or intramuscular (G) prime or prime-boost vaccination with Galsomes containing the indicated mRNA. In B–G, the light and dark blue box plots show the results of mice treated with moIi80tOVA and tNGFR (OVA) mRNA or with moIi80tOVA and survivin (Survivin) mRNA containing Galsomes respectively. The horizontal line shows the mean, while each dot represents one data point or mouse (n=5 for prime, n=5 for prime-boost). Statistical significance was determined using the one-way ANOVA test (Tukey post hoc); two-sided p values are reported. ANOVA, analysis of variance; iNKT, invariant natural killer T cell; mRNA, messenger RNA; OVA, ovalbumin; PBS, phosphate-buffered saline.
Selection of high-confidence MM immunopeptides with clinical relevance
To identify alternative MM antigens for vaccination, we performed imPep using the mouse 5T33MMvt and 5TGM1 cell lines, which are both derived from 5T33MM cells.25 To that end, peptides were eluted and analyzed via LC-MS/MS from MHC-I (H2-Kb and H2-Db, Qa-2)/peptide complexes that were immunoprecipitated from lysates of 5T33MMvt and 5TGM1 cells using the M1/42.3.9.8 antibody (figure 4A).21 MS2 spectra were searched by PEAKS Studio (V.11) against the mouse proteins in the UniProt database, with duplicate analyses performed for both cell lines.35–37 All peptides are available in online supplemental files 1–7. Shared peptides between the two replicates were used to plot the peptide length distribution, showing that immunopeptides dominantly have a length of eight or nine amino acids (figure 4B). These peptides with a length of eight or nine amino acids were considered for GIBBS clustering analysis (figure 4C).38 The results of the complete GIBBS clustering analysis are shown in the supplementary figure for the octamers and nonamers of 5T33MMvt (online supplemental figure 2A,B) and 5TGM1 (online supplemental figure 2C,D). The GIBBS clustering analysis revealed the expected motif for the H2-Db, H2-Kb and Qa-2 alleles for either octamer or nonamer peptides and for both cell lines (figure 4D). The anchor residues for each motif were clearly resolved for the three alleles. Peptide binding prediction with NetMHCpan23 also shows a high proportion of strong binders for both cell lines (figure 4E). Together this shows that both cell lines exhibit the expected MHC class I peptidome without obvious differences.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material

Identification of MHC-I restricted immunopeptides in two related 5T models. (A) Schematic representation of the experimental set-up. 5T33MMvt and 5TGM1 cells were expanded and lysed. Lysates were incubated with an MHC-I specific antibody for immunoprecipitation of MHC-I/peptide complexes. Peptides were retrieved using acidic elution and analyzed by LC-MS/MS. (B) MS2 spectra were searched by PEAKS Studio (V.11 build 20220327) against a database containing all murine sequences listed in UniProt. Peptides identified in both 5T33MMvt and 5TGM1 cells were plotted for length. Octamer and nonamer peptides were submitted for GIBBS clustering analysis. (C) Experimental sequence logos for H2-Db, H2-Kb and Qa-2. (D) Binding prediction using NetMHCpan for the different alleles and octamer and nonamer immunopeptides for both cell lines. MHC-I, major histocompatibility class I; MM, multiple myeloma; MS, mass spectrometry.
To narrow down the selection of putative antigens for vaccination, shared peptides between the 5TMM cell lines were scrutinized, yielding a list of 2034 MHC-I restricted peptides (figure 5A), mostly octamers and nonamers (figure 5B). To select antigens for vaccination, we applied the following criteria for the antigen-derived peptides: (1) presented by both cell lines, (2) multiple peptides per antigen, (3) immunogenicity and (4) their relevance in MM. Proteins presenting more than three peptides were submitted to the Panther database to determine the origin of the immunopeptides (figure 5C), showing a high number of peptides derived from proteins used in RNA metabolism. The immunogenicity of all nonamers was predicted by the online Immune Epitope Database tool, identifying EF1A1 and PICALM as most immunogenic (figure 5D). Based on these criteria, PICALM and EF1A1 were selected as antigens for therapeutic vaccination. Although HSP60 was not among the top candidates in predicted immunogenicity, it met the first two selection criteria and was included due to its strong relevance in MM, demonstrated by the gene expression data from the TT2 cohort (figure 5E–F). Gene expression data demonstrated that HSP60 is upregulated during disease progression and high expression is associated with a poor prognosis. Additionally, the variable Igκ chain was included as well due to its specificity to plasma cells and its significant relevance in MM.

Selection of MHC-I restricted immunopeptides in related 5TMM models. (A) Peptides identified in both 5T33MMvt and 5TGM1 cells were selected for analysis. (B) Distribution showing the prevalence of octamer and nonamer peptides presented in MHC-I. (C) Various cellular proteins present more than one peptide. (D) Nonamer peptides were submitted to the online IEDB tool to predict immunogenicity. (E–F) Expression of EF1A1, PICALM and HSP60 in patients with MM of the TT2 cohort. (E) The scatter plots show PICALM (left), EF1A1 (middle) and HSP60 (right) expression in BMPC of healthy donors and donors diagnosed with MGUS, SMM or MM. MaxStat analysis was used to calculate the optimal separation of patients based on a cut-off value. Data are log transformed, MAS5-normalized Affymetrix signals with each data point representing one donor (n=12–345). Respective probes: 212,506, 204,892, 200,807. Statistical significance was determined by one-way ANOVA (Tukey post hoc); two-sided p values are reported. (F) Kaplan-Meier curves for patients with MM with low (blue) or high (red) expression levels of PICALM (left), EF1A1 (middle) and HSP60 (right). The log-rank (Mantel-Cox) test was used to analyze survival curves (n=345). ANOVA, analysis of variance; BMPC, bone marrow plasma cells; IEDB, Immune Epitope Database; MGUS, monoclonal gammopathy of unknown significance; MHC-I, major histocompatibility class I; MM, multiple myeloma; OS, overall survival; SMM, smoldering MM; TT2, Total Therapy 2.
Intramuscular vaccination with Galsomes containing EF1A1, PICALM, HSP60 and variable Igκ chain mRNA in combination with anti-CD40 antibody therapy impacts MM progression
The therapeutic effects of the imPep-based mRNA vaccine were studied in two different MM models (online supplemental figure 3A). In the aggressive 5T33MM model, mice were vaccinated early by intramuscular Galsome injection. In the slower 5TGM1 model, vaccination was performed after disease establishment. The Galsomes contained mRNA encoding PICALM, EF1A1, HSP60 and the variable Igκ chain (all 0.625 µg), collectively referred to as the imPep mRNA vaccine. Its efficacy was evaluated by measuring serum M-protein levels as a marker for MM disease progression. Galsome vaccination resulted in a decrease in disease levels only in 5TGM1 mice, while vaccinated 5T33MM mice showed no therapeutic benefit (online supplemental figure 3B).
We hypothesized that the therapy efficacy could be enhanced by using a CD40 agonist (αCD40), given the impact of this immunotherapy on various immune-related processes, including DC maturation and T cell stimulation.39 Mice were divided into four groups: PBS, imPep mRNA Galsome vaccine, αCD40 and a combination treatment of the imPep mRNA Galsome vaccine and αCD40 (figure 6A). Mice received an intramuscular injection of PBS or Galsomes on day 3 and 10 (5T33MM) or day 12 and 19 (5TGM1), while αCD40 was administered intraperitoneally on day 7 (5T33MM) or day 16 (5TGM1). To facilitate comparison between Galsome vaccine, αCD40 and combination therapy, results were normalized to the PBS group. We observed the lowest M-protein serum levels in mice treated with the combination therapy (figure 6B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Intramuscular vaccination with Galsomes containing imPep mRNA combined with agonist CD40 therapy reduces MM burden and impacts immune activation. (A) Schematic representation of the experimental set-up. C57BL/KalwRij mice were inoculated with 5T33MM or 5TGM1 cells on day 0. Mice were randomly assigned to one of four groups. On the indicated days, mice in groups 1, 2 and 4 received an intramuscular injection with PBS (group 1) or imPep mRNA containing Galsomes (prime-boost regimen, groups 2 and 4). Mice assigned to groups 3 or 4, received an IP injection with a CD40 agonist (αCD40). Mice were sacrificed when the first mouse reached humane endpoints. Data of groups 2, 3 and 4 were normalized to PBS. (B) Box plot showing the M-protein serum levels of mice treated with imPep mRNA Galsomes, αCD40 or the combination thereof relative to PBS. (C) Representative plots of degranulation marker CD107a and PD-1 expression on CD8+ T cells (left) and box plot graphs showing the relative values (right). (D) Representative plots of degranulation marker CD107a and PD-1 expression on iNKT cells (left) and box plot graphs showing the relative values (right). The light blue, gray and dark blue box plots show the results of mice treated with imPep mRNA Galsomes, αCD40 or the combination thereof respectively. The horizontal line shows the mean, while each dot represents one data point or mouse (n=9 5T33 MM, n=8 5TGM1). Statistical significance was determined using the one-way ANOVA test (Tukey post hoc); two-sided p values are reported. ANOVA, analysis of variance; imPep, immunopeptidomics; iNKT, invariant natural killer T cell; IP, intraperitoneal; MM, multiple myeloma; mRNA, messenger RNA; PBS, phosphate-buffered saline; PD-1, programmed death-1.
To understand the interplay between imPep mRNA Galsome vaccination and αCD40 therapy, we first studied the impact on expression of the antigen-presenting H2-kb molecule on MM cells in BM (online supplemental figure 4A) and spleen (online supplemental figure 4B), showing a statistically significant increase following αCD40 therapy and combination therapy compared with imPep mRNA Galsome vaccination in 5T33MM mice. Given the more pronounced difference in BM, we next assessed the effect of the treatment regimens on professional APCs, potentially involved in T cell activation by imPep mRNA Galsome vaccination. We observed a statistically significant increase in mature DCs and MHC-IIhigh macrophages within the BM after combination therapy compared with imPep mRNA Galsome vaccination and αCD40 monotherapy (online supplemental figure 4C,D). In 5TGM1, Galsome vaccination alone was already sufficient to remodel the immune microenvironment towards a more immunogenic and anti-tumoral state.
We finally addressed the impact of the treatment regimens on NK, iNKT and CD8+ T cells, choosing the spleen for analysis as these cells need to exert their function in the periphery. While αCD40 treatment in monotherapy or combination results in a significant increase in CD8+ T cells, Galsome-treated mice showed impaired levels of iNKT cell numbers in both models (online supplemental figure 4E,F). The cytotoxic effector function of iNKT and CD8+ T cells was studied by measuring the degranulation marker CD107a, while activation and subsequent risk of exhaustion was studied by measuring the expression of the inhibitory receptor PD-1. We observed a statistically significant increase in CD107a+ and PD-1+ CD8+ T cells in the combination treatment, compared with both monotherapies (figure 6C). We further addressed the expression of CD107a+ and PD-1 on iNKT cells, showing that CD107a and PD-1 expression was significantly elevated in mice receiving Galsomes alone or in combination treatment, compared with αCD40 alone (figure 6D). Lastly, NK cell numbers, CD107a and PD-1 levels followed the same pattern as iNKT cells, however less pronounced (online supplemental figure 4G,H). These data show that Galsome vaccination combined with a CD40 agonist is able to activate cytolytic immune cells that potentially can recognize and kill MM cells.
Discussion
We studied for the first time therapeutic mRNA vaccination in MM. We prime-boost vaccinated with Galsomes, LNPs containing αGC and antigen mRNA to activate iNKT and CD8+ T cells, using these Galsomes as a monotherapy or combined with a CD40 agonist in MM. We identified clinically relevant vaccine antigens using imPep, a cutting-edge omics technology, in the 5T33MM preclinical MM model. The preclinical data advocate intramuscular vaccine administration and advise against using survivin, while imPep-identified antigens have potential as vaccine antigens. These data further emphasize the need for combination therapy, resulting in reduced tumor burden, increased MM cell antigenicity and anti-MM immunity.
Most vaccines focus on activation of CD4+ and CD8+ T cells. CD4+ T helper 1 (TH1) cells support antitumor immunity by licensing DCs to become more proficient CD8+ T cell activating cells, by providing direct help to CD8+ T cells to become cytotoxic cells and by exerting tumor cytolysis of MHC-II+ tumors.40 Stimulation of CD4+ TH1 cells following mRNA-based vaccines requires tumor antigen engineering with signal and class II trafficking domains for which several options are available.41 Still, generating CD4+ TH1 responses can be challenging due to the scarcity of specific naive CD4+ T cells and a decline in T cell diversity with age,42 which is particularly relevant in the aging MM population.43 This is exemplified by a study, showing that patients with MM compared with healthy donors possessed fewer CD4+ T cells that react to survivin, one of the antigens used in this study.44 Therefore, we used αGC as a helper molecule, activating iNKT cells that take over the role of CD4+ TH1 cells. iNKT cells license DCs, resulting in attraction and activation of naive CD8+ T cells.45 46 This depends on peptide and glycolipid presentation by the same DC and on the interaction of CD40L expressed on iNKT cells with CD40 expressed on DC.47 Moreover, IFN-γ, produced at high levels by iNKT cells following Galsome administration, supports CD8+ T cells directly.48 iNKT cells also assist in the lysis of MM cells49 50 and, like CD4+ TH1 cells51 can activate NK cells, which can assist in lysis of tumor cells that downregulate MHC-I in order to hide from CD8+ T cells.52 Therefore, we first focused on the role of iNKT cells postvaccination, prompted by the observation of numeric loss following prime-boost intravenous vaccination in MM-bearing mice. This anergic state aligns with previous findings on repeated αGC administration.53–55 However, the nature of iNKT cell responses is influenced by the route of administration.9 33 To explore whether intramuscular injection could mitigate the observed iNKT cell loss, we compared administering the vaccine both intramuscularly and intravenously in healthy mice. Our results showed that the intramuscular route permitted iNKT cell expansion in healthy mice following prime-boost vaccination, while numeric loss was observed after repeated intravenous administration of the vaccines. This observation conflicted with our findings in MM-bearing mice, where the imPep mRNA vaccination was performed intramuscularly and still resulted in iNKT loss. This discrepancy suggests that the presence of chronic stimulation from MM cells leads to a pronounced anergic state, regardless of the administration route. Moreover, administration of a CD40 agonist as a single agent also resulted in anergic iNKT cells, further indicative of the impact of chronic MM cell stimulation on iNKT cell numbers. In contrast to iNKT cells, CD8+ T cell anergy was observed in intravenously vaccinated mice but not in those receiving the intramuscular injection. Notably, activation of cytolytic NK and CD8+ T cells, defined as CD107a+ cells, following intramuscular prime-boost Galsome vaccination required combined therapy with a CD40 agonist, suggesting that the CD40/CD40L interaction between iNKT cells and MM cells in this preclinical model was insufficient to fully license the APCs, corroborated by the finding that all APCs, including MM cells, DCs and macrophages, showed increased levels of MHC molecules.
To study the activation of CD8+ T cells following prime-boost Galsome vaccination, we searched for antigen candidates in the 5T33MM model with clinical relevance. Survivin was identified as an antigen from previously described microarray data showing high transcript values of the BIRC5 gene that encodes survivin.17 Its clinical relevance, already suggested in literature,56 was corroborated using the TT2 gene expression database. BIRC5 was upregulated during disease progression and high BIRC5 expression correlated with lower overall survival in patients with MM. We vaccinated MM diseased mice with Galsomes encapsulating full-length survivin mRNA. This resulted in significantly lower M-protein levels despite a reduction in survivin-specific CD8+ T cells following boost vaccination compared with mice treated with PBS, which showed detectable levels of survivin-specific CD8+ T cells. Such endogenously occurring survivin-reactive T cells have also been documented in patients with glioma and MM.28 57 To understand this loss of survivin-specific T cells following prime-boost Galsome vaccination, we vaccinated healthy and MM-bearing mice with Galsomes containing ovalbumin with or without survivin mRNA. The inclusion of survivin mRNA resulted in a loss of ovalbumin-specific T cells after intramuscular prime-boost vaccination. Vaccination-induced downregulation in survivin expression levels could explain this loss. Low survivin expression, particularly in the G1 phase, has been associated with increased AICD, leading to T cell apoptosis. Alternatively, survivin is upregulated during T cell activation in the G2/M phase, and the generation of survivin-reactive T cells could result in fratricide. Together, these mechanisms, or one of them, may explain the reduced levels of survivin-specific CD8+T cells following prime-boost vaccination.14 15 58 59 This raises concerns regarding the use of survivin as a vaccine antigen. Therefore we also explored alternative antigens, using imPep. This technology is increasingly used by us and others to identify antigen candidates for cancer,60 61 viral,62 and bacterial63 vaccines. In a recent study we used imPep to identify antigens of the intracellular bacterial pathogen Listeria monocytogenes. Galsome mRNA vaccination resulted in potent T cell responses that provided protection in mice challenged with this bacterium.21 Here, we used this approach to identify immunodominant peptides presented by 5T33MM and 5TGM1 cell lines. By using parameters such as predicted immunogenicity and the number of peptides presented from one antigen, we selected HSP60, Idiotype, PICALM, and EF1A1 as antigens. We demonstrated the clinical significance of HSP60 and PICALM, as high expression of these antigens in patients with MM is associated with lower overall survival, making it less likely that MM cells will downregulate antigen expression in an attempt to evade immune recognition. Though this was not the case for EF1A1 in the TT2 cohort, DepMap analysis (data not shown) revealed a strong dependency (scores ≤1.5) on EF1A1 and HSP60 in human MM cell lines. Prime-boost vaccination with this imPep-based mRNA vaccine significantly reduced MM growth in the 5TGM1 model, while no therapeutic effects were observed in the more aggressive 5T33MM model. We reasoned that Galsome vaccination in 5T33MM did not sufficiently induce the maturation and immunogenicity of DCs, which we remedied by combining the intramuscular prime-boost Galsome vaccine with a CD40 agonist. This improved antigen presentation, DC maturation and increased the level of classically activated, so-called M1 macrophages. This combination treatment resulted in both 5T33MM and 5TGM1 in a more cytotoxic profile of activated NK, iNKT and CD8+ T cells as indicated by an increase in the degranulation marker CD107a, PD-1 and a reduction in tumor burden as measured by M-protein concentration. We used CD107a as a functional marker to identify degranulating cytolytic effector cells. Our data demonstrates that the combination treatment enhances effector T cell function compared with either single treatment. These cytotoxic lymphocytes were further characterized by increased levels of PD-1, a marker upregulated on activation and used by tumor cells to protect themselves against cell-mediated cytolysis.64 This shows that our combination strategy effectively engages both iNKT and CD8+ T cells. These findings provide a rationale for combined immunotherapy in MM, in particular antigen-specific Galsome vaccination with non-specific immune activators, such as CD40 agonists. Future research to improve therapy outcomes could scrutinize antigen immunogenicity, dose of antigen mRNA and its formulation in one or more Galsome vaccines. Also, the timing of CD40 agonist delivery could be studied, while including programmed death-ligand 1 (PD-L1) antagonists could lead to more significant reduction in tumor burden by countering PD-L1 and PD-1 upregulation after Galsome vaccination.11
The current study also has broader implications for patients with MM. Several studies have reported weak cellular responses following COVID-19 mRNA vaccination,44 45 suggesting an overall unfavorable immune status of patients with MM. The preclinical data in this study suggest that therapy with CD40 agonists overall improves the immune status of MM-bearing mice, which could be a viable strategy to improve vaccine efficacy in patients with MM.
In conclusion, we provide the first proof-of-concept that the combination of Galsomes, LNPs containing αGC and antigen mRNA, with a CD40 agonist, has therapeutic merit, reducing tumor burden, increasing MM cell antigenicity and anti-MM immunity.
Supplemental material
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request. The mass spectrometry proteomics data, with identifier PXD060156, have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://proteomecentral.proteomexchange.org/PXD060156). The necessary peptide data is included in supplementary tables. Other data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors acknowledge the contribution of the following technical assistants and core facilities: Petra Roman and Elsy Vaeremans, who performed the cloning, plasmid and mRNA production work at the UMCOR Molecular Biology Core Facility of the Vrije Universiteit Brussel; Carine Seynaeve, who performed lab assistance; Charlotte Van De Walle, who helped with procedures in mice at the Animalarium Core Facility of the Vrije Universiteit Brussel; Angelo Willems, Eddy Impe and Geert Stangé of the Flow Cytometry Core Facility of the Vrije Universiteit Brussel for their advice during the design of antibody panels, the acquisition and analysis of cells, and An Staes of the VIB Proteomics Core, who carried out the LC-MS/MS analysis of the immunopeptidomics samples.
References
Footnotes
Contributors AVdV, FT, CT, KM, SM, EJ, LDB, LF, FI, IL, RV, EM and KB collected and interpreted the data. AVdV, EM and KB have drafted the manuscript. All authors read, edited, and approved the final manuscript. EM and KB share last coauthorship. KB is the guarantor. ChatGPT was used for minor text revisions (grammar checks).
Funding This research was performed with financial support from the Vrije Universiteit Brussel under the strategic research program scheme (SRP48, SRP84), the Ghent University under the concerted research action scheme (BOF21/GOA/033), and a Starting Grant to FI (BOF/STA/202209/011), the Scientific Fund Willy Gepts (WFWG2019), the King Baudouin Foundation-Fund Catharina Weekers (2020-J1811380-217916), the Research Foundation Flanders (G040319N) and Kom op tegen Kanker (Stand up to Cancer): the Flemish cancer society (FAF-C/2018/1213). AVdV, SM, LDB, KDV and RV received personal funding from the Research Foundation Flanders as a travel grant (K110324N) under the predoctoral translational (1S73122N), the predoctoral fundamental (1164918N), the junior postdoctoral (12AN524N) and the senior postdoctoral (12I0921N and 1275023N) grant scheme, respectively. The funders had no role in the study design, data collection, analysis, or interpretation, nor in the writing of the manuscript or the decision to submit it for publication.
Competing interests The authors declare the following competing interest: therapeutic nanoparticles and methods of use thereof is the topic of a patent (WO2020058239A1) on which IL an RV are inventors. None of the authors receive any support or remuneration related to this platform.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.