Article Text

Abstract
Background Sialic acid-binding immunoglobulin-like lectin (Siglec)-15-expressing tumor-associated macrophages (TAMs) drive immunosuppression in the tumor microenvironment (TME), promoting CD8+ T cell exhaustion and limiting immunotherapy efficacy. Both blockade of immune checkpoint molecule Siglec-15 and promotion of granulocyte-macrophage colony-stimulating factor (GM-CSF) have been respectively employed in anticancer immunotherapy.
Methods Murine CT26 or MC38 cancer cells were used to establish subcutaneous tumor models in BALB/c or C57BL/6 mice. Tumors were treated with anti-Siglec-15 antibody–GM-CSF chimera (anti-S15×GM CSF) or anti-Siglec-15 antibody via intraperitoneal injection. The TME was analyzed by flow cytometry and ELISA for immune cell infiltration and cytokine levels. Biodistribution and half-life of anti-S15×GM CSF were assessed by intravenous injection in tumor-bearing mice, with GM-CSF levels measured by ELISA. Macrophage reprogramming and antigen presentation were evaluated using bone marrow-derived macrophages and human peripheral blood mononuclear cell-derived macrophages treated with anti-S15×GM CSF, followed by flow cytometry and immunofluorescence assays.
Results Here we report that anti-S15×GM CSF displays superior function to suppress the progression of Siglec-15-overexpressing MC38 colon cancer engrafted in mice compared to anti-Siglec-15 antibody or GM-CSF alone. Different from the injected GM-CSF which is distributed broadly in various organs and tissues of mouse, the injected anti-S15×GM CSF is preferentially accumulated in Siglec-15-positive tumor cells and TAMs. Anti-S15×GM CSF not only extends the half-life of GM-CSF in vivo, but also reduces the off-target effect of GM-CSF through TAM-specific delivery. In addition to Siglec-15 blockade, anti-S15×GM CSF effectively reprograms immunosuppressive TAMs to a proinflammatory phenotype, enhancing antigen presentation by macrophages to activate T cells.
Conclusions In summary, our results reveal that anti-S15×GM CSF may serve as an effective therapeutic approach for solid tumors.
- Antibody
- Immunotherapy
Data availability statement
No data are available. Data is included within the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Sialic acid-binding immunoglobulin-like lectin (Siglec)-15, an immune checkpoint molecule, is highly expressed on tumor cells and tumor-associated macrophages (TAMs), suppressing CD8+ T cell activity and promoting immunosuppression. Blocking Siglec-15 (eg, with NC318 antibody) shows antitumor potential, but its efficacy is limited. Granulocyte-macrophage colony-stimulating factor (GM-CSF), a cytokine that activates macrophages and dendritic cells, has antitumor effects at high doses. Antibody-cytokine fusion strategies have been explored to improve cytokine targeting and reduce systemic toxicity.
WHAT THIS STUDY ADDS
We have developed anti-S15×GM CSF, a novel fusion protein combining an anti-Siglec-15 antibody with GM-CSF, which simultaneously blocks Siglec-15 and delivers GM-CSF specifically to TAMs. The anti-S15×GM CSF extends GM-CSF’s half-life (from 4.5 hours of free GM-CSF to 15.2 hours) and reprograms immunosuppressive TAMs into proinflammatory M1 macrophages, leading to enhancement of antigen presentation and CD8+ T cell activation. In mouse models, this chimera significantly suppresses tumor growth compared with anti-Siglec-15 antibody alone, demonstrating synergistic effects of Siglec-15 blockade and immune activation of TAMs by GM-CSF.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study provides a blueprint for designing dual-functional immunotherapies that combine checkpoint blockade with cytokine delivery, improving efficacy and safety. It highlights Siglec-15 as a viable target for TAM-focused therapies, potentially informing clinical trials combining anti-Siglec-15 agents with other immunomodulators. The study may also facilitate the development of fusion protein-based therapies to overcome limitations of cytokine monotherapy and improve personalized cancer treatment.
Introduction
The discovery of immune checkpoints has revolutionized cancer immunotherapy.1 2 The sialic acid-binding immunoglobulin-like lectin (Siglec) family of membrane proteins are such immune checkpoints and play a crucial role in the regulation of immune homeostasis.3 Siglec-15, a protein encoded by the SIGLEC-15 gene, is found to be upregulated in tumor cells and tumor-associated macrophages (TAMs), leading to exhaustion of CD8 T cells and severe immunosuppression within the tumor microenvironment (TME).4–6 Blockade of Siglec-15 (such as anti-Siglec-15 antibody NC318) thus shows great promise in tumor immunotherapy.4 7–9 However, the antitumor efficacy of Siglec-15 blockade alone remains unclear.10 11
Cytokines including interleukin (IL)−2, IL-12, IL-15, IL-21, granulocyte-macrophage colony-stimulating factor (GM-CSF), and interferon-α have long been demonstrated to have a beneficial effect on suppressing tumor growth in preclinical mouse cancer models12–14 and clinical studies.15 16 For instance, GM-CSF, an immunomodulatory cytokine stimulating the proliferation and activation of macrophages and dendritic cells (DCs),17–23 has been used in various clinical trials, especially in combination with oncolytic virus24–28 or immune checkpoint inhibitor.29 30 However, like other cytokines, high concentrations of GM-CSF can also cause severe toxicity to healthy cells and tissues.31 Therefore, specifically delivering GM-CSF to tumor sites and reducing its concentration in peripheral bloodstream is the key in GM-CSF-based tumor therapy.
Various approaches have been explored to achieve targeted delivery of GM-CSF to the tumor site,13 including conjugating GM-CSF with drugs or nanoparticles32 or using gene therapy to express GM-CSF directly in tumor cells.33 34 One promising resolution is to combine tumor-specific antibodies with cytokines as fusion proteins.35 These antibody-cytokine chimeras have the advantage of exhibiting enhanced potency, improved pharmacokinetics, and reduced toxicity compared with the individual cytokines.36 37 For example, a fusion protein consisting of IL-2 and a tumor-targeting antibody fragment has been shown to selectively activate T cells within tumors, leading to potent antitumor effects.38 39 Such antibody-cytokine fusion proteins represent a promising class of therapeutics with the potential to selectively target specific cell types including tumor cells or tumor-associated immune cells, thus modulating immune responses in a controlled manner.
In the present study, we develop an effective therapy for solid tumors by taking advantage of the antibody-cytokine fusion protein strategy. In specific, GM-CSF is fused with a functional anti-Siglec-15 antibody to form a chimera of anti-Siglec-15 antibody and GM-CSF (anti-S15×GM CSF). Our results show a strong TAM-targeting capacity of anti-S15×GM CSF and that anti-S15×GM CSF dose-dependently suppresses the progression of tumors engrafted in mice. Mechanistically, anti-S15×GM CSF reprograms TAMs from an immunosuppressive phenotype to a proinflammatory phenotype, as evidenced by enhanced antigen presentation by macrophages to activate specific effector T cells. These findings reveal anti-S15×GM CSF as a promising immunotherapeutic approach for cancer treatment.
Materials and methods
Tissue microarrays and immunostaining
For immunofluorescence staining, human tumor and adjacent normal tissues were formalin-fixed and paraffin-embedded using standard procedures. The sections were immunolabeled with the primary antibodies, including anti-Siglec-15 (clone B4A6, Biosion), anti-CD68 (clone KPI, Thermo Fisher, USA), anti-PD-L1 (clone E1L3N, #13684, CST), respectively. All the immunostaining was performed and analyzed by WiSee Biotechnology (Shanghai, China).
Acquisition of gene expression profiles from datasets
RNA-seq data of tumor tissues and corresponding clinical information in The Cancer Genome Atlas were downloaded through UCSC Xena (http://xena.ucsc.edu). The expression of genes analyzed in normal tissues was collected from the Genome Tissue Expression database. The expression levels of Siglec-15 in different types of tumor and normal tissues were analyzed.
Production of anti-S15×GM-CSF heterodimer
Anti-Siglec-15×GM CSF fusion protein is a bispecific antibody comprizing an antibody targeting Siglec-15 (clone A2A5, Biosion), which is fused to GM-CSF fusion protein at the c-terminal of the heavy chain. Anti-Siglec-15×mouse GM CSF (mGM CSF) fusion protein was generated with the same format as Anti-Siglec-15×GM CSF fusion protein. GM-CSF-FC comprizing a GM-CSF fusion protein fused at the c-terminal of IgG4-Fc and mGM-CSF-FC share the same format with GM-CSF-Fc. The sequences encoding human GM-CSF (hGM-CSF) or mGM-CSF (amino acids from Ala 18 to Glu 144) were designed according to the sequence information in the Uniprot protein database (https://www.uniprot.org/uniprotkb/P04141 or https://www.uniprot.org/uniprotkb/P01587). Recombinant bispecific antibodies with human IgG4 heavy chain and light chain were constructed in the PTT5 vector and then the plasmids containing light chain and heavy chain were co-transfected into Chinese hamster ovary cells (CHO) cells. The genes of recombinant fusion protein were also constructed in the PTT5 vector and then transfected into CHO cells. The supernatant was collected and purified by Protein A beads to obtain proteins for testing.
Construction of Siglec-15 overexpressing cell lines
Lentivirus expressing human or mouse Siglec-15 was used to generate human Siglec-15 overexpressing 293 T cell line or mouse Siglec-15 overexpressing MC38 cell line. Briefly, when cultured cells achieved∼50% confluent, the culture medium was replaced with fresh Dulbecco’s Modified Eagle’s Medium (DMEM) complete medium. The cells were then exposed to the lentivirus along with polybrene (5 µg/mL) for 48 hours. Following transduction, cells were selected in the presence of puromycin (1 µg/mL) for 7 days with daily medium changes. Single colonies were screened through dilution, and gene overexpression in cells was validated using flow cytometry.
Isolation of CD14-positive monocytes and CD3-positive T cells
According to the manufacturer’s instructions, CD3-positive cells were isolated using EasySep Human T Cell Iso Kit (STEMCELL, 17951), and CD14-positive cells were isolated using EasySep Human CD14 Positive Select Kit II (STEMCELL, 17858).
Induction of macrophages to TAMs
To obtain human macrophages, CD14 cells were isolated from human peripheral blood mononuclear cells (PBMCs). After isolation, cells were cultured in 1640 supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 100 ng/mL M-CSF (MCF-H5218, ARCO) for 5 days, with the medium changed every 2 days. To obtain mouse macrophages, the mouse tibia and femur were isolated and flushed with cold phosphate-buffered saline (PBS) through a 25 G needle in a sterile environment. Cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 100 ng/mL M-CSF (51112-MNAH, Sino Biological) for 5 days, with the medium changed every 2 days. To obtain TAMs, 4T-1 (for inducing mouse TAMs) or A549 (for inducing human TAMs) cells were cultured to 75% density, and then washed twice and replaced with fresh culture medium. After 24 hours culture, the conditioned medium was collected. Mouse bone marrow-derived macrophages (BMDMs) or human macrophages were then incubated with the respective conditioned medium for 48 hours to obtain mouse or human TAMs.
Protein labeling
According to the manufacturer’s instructions, we first dissolved the Siglec-15 protein, LRRC4C protein, mGM-CSF, or anti-S15×mGM CSF with PBS buffer (pH 7.2–8.0) in an amine-free buffer (HyClone, SH30028.02). Next, 10 mM biotin (Thermo Fisher, 21217) in reagent solution was mixed with 1–10 mg/mL prepared protein. The mixture was incubated at room temperature for 30 min and used directly afterward.
Characterization of anti-S15×GM-CSF chimera
For detecting Siglec-15 binding capacity, a 96-well microtiter plate was coated with goat anti-human IgG Secondary Ab (109-005-098, Jackson Immune Research) in PBS overnight at 4°C. Anti-Siglec-15×GM CSF or control antibody was then added at different concentrations (orderly diluted from 66.6 nM to 0.004267 nM). Biotin-labeled human Siglec-15 protein (200 ng/well) was added and finally incubated with SA-HRP (016-030-084, Jackson Immune Research). To detect ligand blocking capacity of fusion protein, a 96-well microtiter plate was coated overnight at 4°C with human Siglec-15 (200 ng/well) diluted in PBS. The microtiter plate was titrated with anti-Siglec-15×GM CSF or control antibody in a similar manner. Biotin-labeled human LRRC4C protein (200 ng/well) was then added and finally incubated with SA-HRP (016-030-084, Jackson Immune Research). After incubation with SA-HRP, TMB (Huzhou InnoReagents, TMB-S-002) was added and the absorbance at 450–630 nm was measured using a microplate reader. To detect the binding of antibody binding to cell surface Siglec-15, HEK293T cells overexpressing human Siglec-15 or MC38 cells overexpressing mouse Siglec-15 were cultured in 96-well plates at a density of approximately 10,000 cells per well. Anti-Siglec-15×GM CSF or control antibody were added at different concentrations (diluted from 66.6 nM to 0.004267 nM) to incubate with cells. The fluorescent signal was detected by flow cytometry following incubation with fluorophore-conjugated secondary antibody. To detect GM-CSF or mGM-CSF biofunctional, TF1 or FDC-P1 cells (1×105/well) were seeded into cell culture plates overnight. Anti-Siglec-15×GM CSF or GM-CSF at different concentrations were added to cells and incubated for 48 hours. Finally, Lite V.2.0 luminescent cell viability reagent (Vazyme Biotech) was added to count the cells.
Flow cytometry
Fluorescence-activated cell sorting (FACS) analysis was performed using a Kaluza flow cytometer (Beckman Coulter, Miami, USA) and the data was analyzed with FlowJo V.10.0 software. Samples were staining involved FVS510 (564406, BD Biosciences), mixed with FACS buffer containing mouse TruStain Fc block (422302, BioLegend) or human TruStain Fc block (156604, BioLegend) before antibody stain. Surface antigens were then stained for 30 min on ice. For intracellular staining, Transcription Factor Buffer Set (562574, BD Biosciences) was used in accordance with the manufacturer’s instructions. Unless otherwise specified, cell populations were defined by the following markers for humans: FITC-CD80 (clone W17149D, 305206), PerCP-Cy5.5-CD163 (clone GHI/61, 333608), Pe-Cy7-CD4 (clone A161A1, 357410), PerCP/Cyanine5.5, APC-CD8 (clone SK1, 344722), FITC-HLA-A2 (clone BB7.2, 343304) and FITC-HLA-DR (clone L243, 307606) were purchased from BioLegend. For mouse: FITC-anti-CD45 (clone 30-F1, 553079), BV605-anti-CD11b (clone M1/70, 563015), PE-Anti-F4/80 (clone T45-2342, 565410), BV421-Anti-CD86 (clone GL1, 564198), Alexa Fluor 647-anti-CD206 (clone MR5D3, 565250), PE-Cy7-anti-CD11c (clone HL3, 558079), and PerCP-Cy5.5-anti-I-A/I-E (clone M5/114.15.2, 562363). APC-Cy7-anti-CD45 (clone 30-F11, 557659), FITC-anti-CD3e (clone 145–2 C11, 553061), PerCP-Cy5.5-anti-CD4 (clone RM4-5, 550954), PE-Cy7-anti-CD8a (clone 53–6.7, 552877), were purchased from BD Biosciences and PerCP/Cyanine5.5-anti-H-2Kb (clone AF6-88.5, 116516) were purchased from BioLegend. The tissues were cut into small cubes of 1 mm3 and digested with Collagenase Type I (1 mg/mL, Thermo Fisher, 17100017), Collagenase Type II (1 mg/mL, Thermo Fisher, 17101015), Collagenase Type IV (1 mg/mL, Thermo Fisher, 17104019) and DNase I (1 U/mL, Sigma-Aldrich, D5025) in Roswell Park Memorial Institute 1640 medium for 30 min at 37℃.
Binding of anti-S15×GM-CSF to target cells
To validate the preferential binding of anti-S15×mGM CSF to Siglec-15-positive cancer cells, MC38 cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) and mixed with non-labeled MC38 cells overexpressing Siglec-15. To validate the preferential binding of anti-S15×mGM CSF (10 µg/mL) to Siglec-15-positive TAMs, MC38 cells were labeled with CFSE and then mixed with non-labeled TAMs. To validate the preferential binding of anti-S15×mGM CSF (10 µg/mL) to Siglec-15-positive TAMs, not GM-CSF receptor (CSFR)-positive cells, FDC-P1 cells were labeled with CFSE and then mixed with non-labeled TAMs. Cells were labeled with PE-conjugated anti-S15×mGM CSF and isotype-matched control and then analyzed by flow cytometry.
Antigen presentation by macrophages
Macrophages were induced as described above. To detect the ability of macrophages to ingest, process, or present antigens, macrophages were incubated with FITC-dextran (1 mg/mL, Mr=40 000, FD40S, Sigma), DQ-OVA (20 µg/mL, D12053, Invitrogen) or OVA257-264 (25 µg/mL, # HY-P1489A, MCE) peptide for 1 or 2 hours in the presence or absence of equal amounts of GM-CSF (79.8 ng/mL) or anti-S15×mGM CSF (173.9 ng/mL). Antigen uptake was quantified based on the mean fluorescence intensity.40 For antigen presentation analysis, macrophages were surface stained with PE-anti-mouse H-2Kb bound to SIINFEKL antibody (clone 25-D1.16, 141604, BioLegend).41
Siglec-15 binds to T cells and T cell proliferation assay
For Siglec-15 binding to T cells, CD3 (1 µg/mL) (clone OTK3, 14-0037-82, Thermo Fisher)/CD28 (1 µg/mL) (clone CD28.2, 16-0289-81, Thermo Fisher) antibody incubated with T cells for 3 days. Cells (1×106) were then incubated with 0.3 mL of Neuraminidase A (P0722, NEB) at 37°C for 1 hour for removing sialic acid. Siglec-15-FC (10 µg/mL) (10 µg/mL HER2-FC served as negative control) was used to detect Siglec-15 binding to T cells. For the T cell proliferation assay, human PBMCs were resuspended in PBS and mixed with CFSE (1 µM, C34554, Thermo Fisher) in equal volume (cell density 1×106 /mL). According to the experimental requirements, T cells loaded with CSFE were incubated with anti-CD3 (1 µg/mL) and anti-CD28 (1 µg/mL) antibodies in the presence of or absence of recombinant human Siglec-15 protein and different antibodies for 3 days at 37°C. Cells were analyzed by flow cytometry.42
Real-time quantitative PCR
To detect macrophage messenger RNA (mRNA) level changes, total RNA was extracted by using the FastPure Cell/Tissue Total RNA Isolation Kit V2 (RC112-01, Vazyme Biotech) according to the manufacturer’s instructions. Quantitative PCR was performed using SYBR Green (Q221-01, Vazyme Biotech) on an Applied Biosystems 7300 Real-Time PCR (Thermo Fisher), and data were normalized by the level of GAPDH expression in each individual sample. The 2(−ΔΔCT) method was used to calculate relative expression changes. The primer sequences were as follows: IL-6 (sense), ACTCACCTCTTCAGAACGAATTG; IL-6 (antisense), CCATCTTTGG AAGGTTCAGGTTG; CD163 (sense), AAAAAGCCACAACAGGTCGC; CD163 (antisense), CTTGAGGAAACTGCAAGCCG; Arg-1 (sense), CTTGGCAAAAGACTTATCCTTAG; Arg-1 (antisense), ATGACATGGACACATAGTACCTTTC; IL-10 (sense), TCAAGGCGCATGTGAACTCC; IL-10 (antisense), GATGTCAAACTCACTCATGGCT.
Western blot
Proteins extracted from cell or tissue lysis were carefully fractionated using sodium dodecyl sulfate-ployarylamide gel electrophoresis (SDS-PAGE) with either 10% or 12% acrylamide concentrations, operated within a suitable running buffer. Subsequently, these resolved proteins were efficiently transferred onto polyvinylidene fluoride (PVDF) (1620177, Bio-Rad) membranes through the application of a reliable wet transfer technique. The membranes were then blocked with blocking solution for 30 min and then incubated with primary antibodies against Siglec-15 or GAPDH (60004–1-Ig, Proteintech), followed by three washes and then incubation with HRP-conjugated anti-mouse IgG (H+L) (315-035-045, Jackson Immune Research). Uncropped data of Western blot were presented online supplemental file 1.
Supplemental material
In vivo half-life, biodistribution, and antitumor efficiency of anti-S15×mGM-CSF
All animal studies were carried out following protocols approved by the Animal Ethical and Welfare group PKU-Nanjing Joint Institute of Translational Medicine, Raygen Health Molecular Medicine Technology (Approval no. IACUC-2021–051). C57BL/6 mice (6–8 weeks) were purchased from the Vital River Laboratory Animal Technology. To compare the half-life of anti-S15×mGM CSF with mGM-CSF, mice were intravenously administered with mGM-CSF-biotin (4.79 mg/kg) or anti-S15×mGM-CSF-biotin (10.43 mg/kg) at equivalent molar mass. Mouse serum was collected at various time points and analyzed by ELISA. For the ELISA assay, a 96-well microtiter plate was coated with anti-GM-CSF antibody (sc-377039, Santa Cruz) (1 µg/well) overnight at 4°C. The binding of anti-S15×mGM-CSF-biotin, mGM-CSF-biotin or serum to the microtiter plate was detected by SA-HRP (016-030-084, Jackson Immune Research). To establish the animal model, 3×106 MC38-mS15 cells were subcutaneously injected into the right flank of male C57BL/6 mice (6–8 weeks old). The tumor-bearing mice were grouped at random to study the biodistribution and tumor targeting ability in vivo. To evaluate the biodistribution of the therapeutic reagents, mice inoculated with MC38-mS15 cells were intraperitoneally (i.p.) injected with anti-S15×mGM-CSF-Bio (10.43 mg/kg) or an equal amount of mGM-CSF-Bio (4.79 mg/kg) on day 14 postinoculation. Four days post-treatment, tumors and major organs were harvested and analyzed using an ELISA assay to quantify the distribution of the reagents. For the ELISA assay, a 96-well microtiter plate was coated with mGM-CSFR-His (1 µg/well) overnight at 4°C. The binding of anti-S15×mGM CSF, mGM-CSF-Fc or tissue lysis to the microtiter plate was detected by goat anti-human IgG-Fc HRP antibody (109-036-098, Jackson Immune Research) followed by color development and measurement at 450–630 nm using a microplate reader.43 To evaluate the antitumor effects of the reagents, 3×106 MC38-mS15 or 1×106 CT26 cells were subcutaneously injected into the right flank of male C57BL/6 mice (6–8 weeks old) or BALB/c mice (6–8 weeks old), respectively. Mice were randomly assigned to different groups and i.p. injected with anti-S15×mGM CSF or other antibodies once every 2 days for a total of 5 injections from day 7 post-tumor inoculation. During treatments, tumor volume and body weight were monitored carefully. Uncropped data of tumor photo were presented in online supplemental file 1. To calculate the tumor volume, the following formula was used: V=L (length) × W2 (width)/2. To detect the preferential delivery of GM-CSF to Siglec-15-positive cells, after i.p. injection of the same dose of GM-CSF into 4T-1 tumor-bearing mice, tumor tissues were isolated after 48 hours. Follow the tumor dissociation, single-cell suspensions were stained with F4/80 (for macrophage labeling), CD11c (for DC labeling) and Alexa Fluor 647-conjugated AffiniPure Goat Anti-Human IgG (109-605-190, Jackson Immune Research). After extensive washing, cells were analyzed by flow cytometry.
Cytokine detection
The levels of IL-10, TGF-β, IL-6 and TNF-α in tumor or serum were tested using ELISA kits (Liankebio) according to the manufacturer’s protocols. To determine the cytokine in vivo, tumor samples were harvested, homogenized with radioimmunoprecipitation assay (RIPA) buffer (4°C) and centrifuged to collect the supernatant. All samples were diluted 1:1 in diluent for ELISA sets and measured in triplicate. After extensive washing, the absorbance (450 nm) was read by an automated microplate reader.
Statistical analysis
All experimental data were calculated as mean±SD. The variation among the test groups was quantified using the paired, two-tailed Student’s t-test, one-way analysis of variance (ANOVA) and two-way ANOVA for repeated measurements with Tukey’s post hoc comparisons. Data analysis for the animal studies involved two-way ANOVA with Sidak’s multiple comparison tests. Statistical significance for all data sets was set as p<0.05 (GraphPad Prism Software). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. Statistical data were presented in online supplemental file 2.
Supplemental material
Results
TAMs express high levels of Siglec-15
To test the feasibility of specific delivery of GM-CSF to TAMs by targeting the immune checkpoint molecule Siglec-15, we first validated the expression pattern of Siglec-15. In line with the previous reports,9 44 Siglec-15 was heavily expressed in various human tumor types (figure 1A–C). In the murine cancer model, Siglec-15 level in CT26 colon cancer was significantly higher than that in non-tumor organs (figure 1D, left). The Siglec-15 expression in macrophages under various conditions was also examined. Following the differentiation of monocytes into macrophages by GM-CSF and M-CSF treatment, the Siglec-15 level was markedly increased (figure 1D, right). Double immunofluorescence labeling of lung adenocarcinoma (LUAD)and adjacent normal tissue (Normal) sections further showed the upregulation of Siglec-15 expression in tumor cells or CD68-positive TAMs (figure 1E,F). Siglec-15 level in macrophages was also markedly increased when treated with tumor cell conditional culture medium which induced macrophages to TAMs (figure 1G). The results strongly demonstrate that Siglec-15, which predominantly expresses in TAMs and tumor cells, is an ideal target for selective delivery of cytokines to TAMs or tumor cells.

Tumor sites particularly TAMs express high levels of Siglec-15. (A) Representative images of immunohistochemical (IHC) staining for Siglec-15 and PD-L1 are presented from human head and neck cancer (n=93) and human lung cancer (n=150) microarrays. Scale bar, 100 µm. (B) A statistical graph representing microarray. (C) The mRNA expression levels of SIGLEC-15 in 4 types of human cancers and corresponding normal tissues were assessed by meta-analysis. (D) Left, qPCR shows Siglec-15 mRNA expression in different tissue sites in CT26 tumor-bearing mice (n=6). Right, qPCR shows Siglec-15 mRNA expression on human monocytes and macrophages induced by M-CSF (100 n g/mL) or GM-CSF (20 ng/mL) (n=3). (E) Representative images of IHC staining of CD68 and Siglec-15 in the microarray of human lung adenocarcinoma (LUAD) and adjacent normal tissue (normal). Scale bar, 50 µm. (F) Representative histograms of Siglec-15 expression on macrophages, compared with the stain with isotype control antibody (n=3). (G) Western blot analysis of Siglec-15 expression in macrophages induced by different conditions. Left, represents human macrophages. Right, represents mouse macrophages. *p<0.05, **p<0.005, ***p<0.001, ****p<0.0001 versus the indicated. P values were determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test (D) or unpaired t-test (F). BMDM, bone marrow-derived macrophage; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; mRNA, messenger RNA; PD-L1, programmed death-ligand 1; qPCR, quantitative PCR; Siglec, sialic acid-binding immunoglobulin-like lectin; TAMs, tumor-associated macrophages; DAPI, 4',6-diamidino-2-phenylindole.
Anti-S15×GM-CSF production and molecular characterization
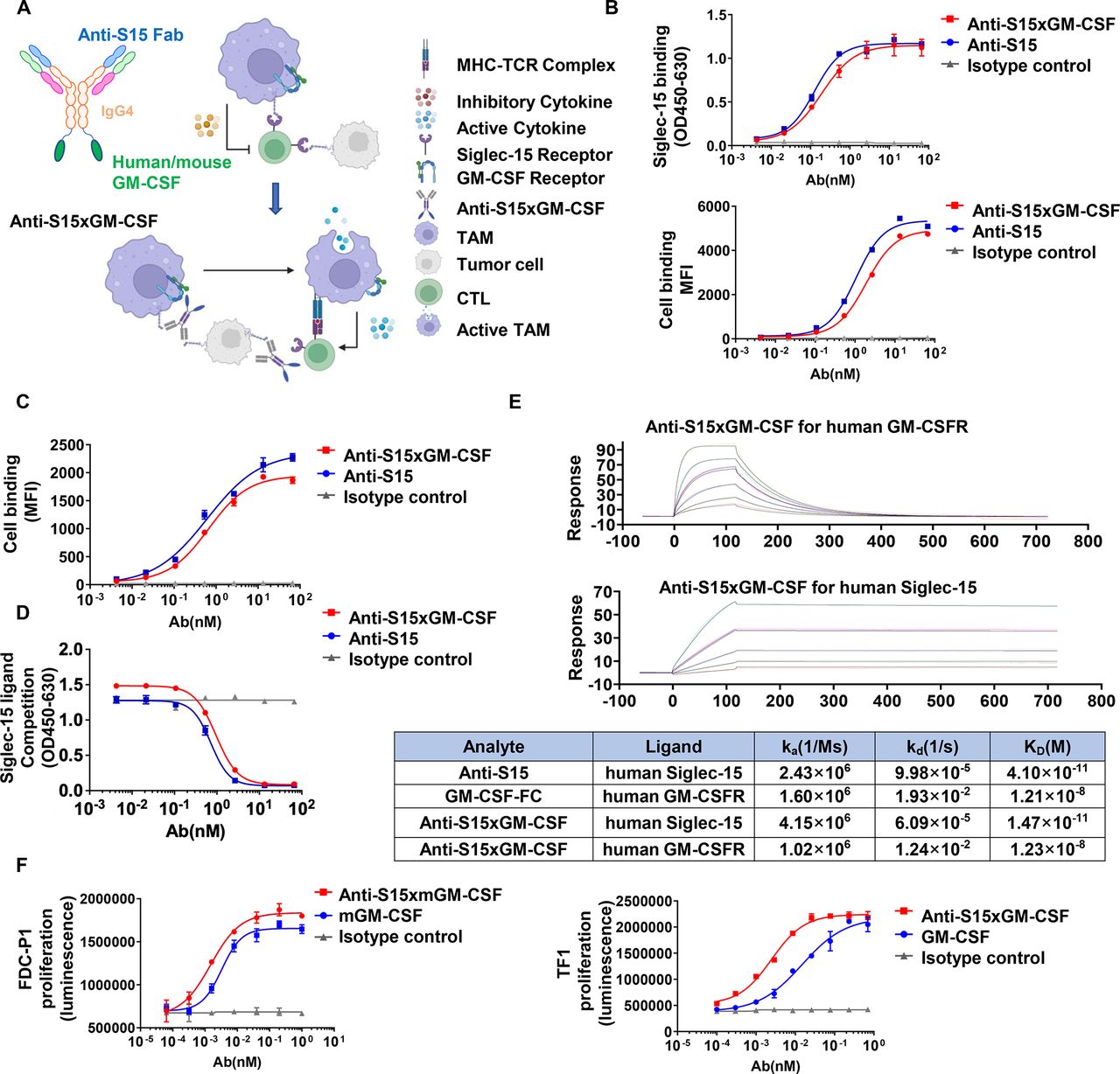
To take advantage of the predominant expression of Siglec-15 on TAMs, we designed an anti-S15×GM CSF chimera to ensure both the blockade of the interaction between TAM Siglec-15 and undefined lymphocyte Siglec-15 counter-receptor, and the specific delivery of GM-CSF to TAMs. Anti-Siglec-15 monoclonal antibody (clone A2A5 or BSI-060T) that displayed high affinity to human-Siglec-15 and mouse-Siglec-15, was capable of strongly reversing Siglec-15-mediated T cell suppression in vitro.45 Through antibody engineering, hGM-CSF or mGM-CSF was fused to the Fc C-termini of the anti-Siglec-15 antibody (figure 2A). To test whether the anti-S15×GM CSF chimera retains the biological characteristics of both anti-Siglec-15 antibody and GM-CSF, a capture ELISA was performed. Anti-S15×hGM CSF displayed high binding capacity to human Siglec-15 (figure 2B, top). To test the binding of Anti-S15 to Siglec-15, we generated 293 T cells overexpressing human Siglec-15 and MC38 cells overexpressing mouse Siglec-15 (online supplemental figure S1A, B), respectively. In 293 T cells overexpressed with human Siglec-15, higher binding of anti-S15×GM CSF to cell surface Siglec-15 was also detected by flow cytometry analysis (figure 2B, bottom). Similarly, the binding of anti-S15×mGM CSF to murine Siglec-15-expressing MC38 cells was also detected by flow cytometry analysis (figure 2C). We also validated the binding of anti-S15 or anti-S15×mGM CSF to BMDM or human macrophage throughflow cytometry analysis (online supplemental figure S2A, B).
Supplemental material

Design and molecular characterization of anti-S15×GM CSF. (A) Schematic of the anti-Siglec-15×GM CSF chimera showing the anti-Siglec-15 region (cross with human and mouse), GM-CSF region or mGM-CSF, and hIgG4 Fc region. The image was created using BioRender.com. (B) Top, binding of anti-S15×GM CSF and anti-S15 antibody to Siglec-15. Bottom, binding of anti-S15×GM CSF and anti-S15 antibody alone to 293T-Siglec-15-overexpress cells. (C) Binding of anti-S15×GM CSF and anti-S15 antibody to MC38-mSiglec-15-overexpress cells. (D) Blockade of Siglec-15 ligand LRRC4C binding to Siglec-15 by anti-S15×GM CSF or anti-S15 antibody. (E) Kinetic association (Ka), dissociation (Kd) and affinity (KD) for binding of the indicated molecules to human Siglec-15 or GM-CSFR measured by surface plasmon resonance. (F) Proliferation curve of human TF-1 and mouse FDC-P1 cells. GM-CSFR, granulocyte-macrophage colony-stimulating factor receptor; MFI, mean fluorescence intensity; MHC, major histocompatibility complex; Siglec, sialic acid-binding immunoglobulin-like lectin; TAMs, tumor-associated macrophages; CTL, Cytotoxic T lymphocytes; TCR, T cell receptor.
LRRC4C and CD11b have been identified as ligands for Siglec-15 previously.46 47 In particular, Lenza et al reported that CD11b mediated the interaction between Siglec-15 and primary T cells implicating that T cells might also express counter-receptor for Siglec-15. We found that the binding of Siglec-15 to T cell surface was time-dependently increased following T cell activation by CD3/CD28 antibodies (online supplemental figure S3A). The increase of Siglec-15 binding to T cells following T cell activation is similar to the upregulation of PD-1 and TIGIT, two markers of T cell exhaustion.48 49 Supporting the inhibitory role of potential Siglec-15 counter-receptor in T cell proliferation, soluble Siglec-15 doses dependently suppressed the proliferation of both CD8 and CD4 T cells (online supplemental figure S3B). Previous work by Sun et al47 reported that LRRC4C may serve as a counter-receptor for tumor cell Siglec-15. We then performed a ligand blocking assay to test whether anti-S15×GM CSF can affect the LRRC4C-Siglec-15 binding. As shown in figure 2D, anti-S15×GM CSF markedly blocked the binding of LRRC4C to Siglec-15, suggesting the potential Siglec-15-blocking function of anti-S15×GM CSF. Interestingly, anti-S15×GM CSF chimera exhibited a roughly hundreds higher binding affinity to Siglec-15 than to GM-CSFR (figure 2E). To test whether anti-S15×GM CSF retains GM-CSF function, human TF-1 cells and mouse FDC-P1 cells were used to examine the activation of GM-CSFR by anti-S15×hGM CSF or anti-S15×mGM CSF. As shown in figure 2F, anti-S15×hGM CSF, and anti-S15×mGM CSF strongly promoted the proliferation of TF-1 and FDC-P1 cells, respectively. This may provide anti-S15×GM CSF with an advantage in preferentially directing GM-CSF to Siglec-15-expressing cells or macrophages. To test this, we mixed CFSE-labeled MC38 cells with non-labeled MC38-mS15 or TAMs and measured their binding to anti-S15×GM CSF and other antibodies. The results showed preferential directing GM-CSF to Siglec-15-expressing MC38 cells or TAMs by anti-S15×mGM CSF (online supplemental figure S4A, B). In a similar manner, we mixed CFSE-labeled FDC-P1 cells with TAMs and measured the binding of GM-CSF. The anti-S15×mGM CSF was found to direct more GM-CSF to Siglec-15-expressing TAMs than GM-CSFR-expressed cells (online supplemental figure S4C). To further investigate how anti-S15×GM CSF directs GM-CSF to Siglec-15 positive cells in vivo, we perform mice inoculated with 4T-1 tumors, and i.p. injection of equal amount of IgG4-FC (3.08 mg/kg), mGM-CSF-FC (4.79 mg/kg) or anti-S15×GM CSF (10.43 mg/kg) on day 7. Tumor single-cell suspensions were collected after 1 day, and subjected to antibody staining and flow cytometry analysis. The results showed anti-S15×mGM CSF not mGM-CSF preferentially directing GM-CSF to TAMs and DCs (online supplemental figure S4D). Taken together, these results demonstrate that the anti-S15×GM CSF chimera not only recapitulates the dual function of anti-Siglec-15 antibody and GM-CSF, but also improves TAM-specific targeting of GM-CSF.
Immunomodulatory effects of anti-S15×GM-CSF
Previous studies have established that through ligating Siglec-15 counter-receptor on T cell surface, Siglec-15 on TAM or tumor cell surface can inhibit T cell proliferation and activation.4 To study the effect of anti-S15×GM CSF on the blockade of Siglec-15-mediated T cell inhibition, we monitored the T cell Siglec-15 binding and proliferation induced by ligation of CD3 and CD28.50 To test whether anti-S15×GM CSF affects Siglec-15 binding to T cells, we treated T cells with sialidase and used it as a negative control (figure 3A). To test T cell proliferation, CFSE-labeled T cells were activated by antibodies against CD3 and CD28 in the presence or absence of anti-S15×GM CSF for 3 days. The proliferation of T cells was assessed by flow cytometry using CD4 and CD8 as phenotype markers (figure 3B). The results indicated that anti-S15×GM CSF effectively eliminated the Siglec-15-mediated inhibition of T cell function, highlighting its potential in tumor immunotherapy.

Anti-S15×GM CSF potently abolishes the inhibitory effect of Siglec-15 on T cell function. (A) CD3 T cells were isolated from human PBMCs. After 3 days of stimulation with anti-CD3 and anti-CD28, PE-Cy7 was used to label CD8, APC was used to label CD4, and FITC was used to label Siglec-15-FC (HER2-FC as negative control) (n= 3). (B) Representative percentages of carboxyfluorescein succinimidyl ester (CFSE)-labeled human peripheral CD8+ T cells after indicated treatments (n=3). ****p<0.0001 versus indicated. P values were determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test (A,B). GM-CSFR, granulocyte-macrophage colony-stimulating factor receptor; PBMC, peripheral blood mononuclear cell; Siglec, sialic acid-binding immunoglobulin-like lectin; APC, allophycocyanin; FITC, fluorescein isothiocyanate.
As a potent cytokine, GM-CSF plays a vital role in regulating the function and development of a wide range of immune cells, including granulocytes, monocytes/macrophages, and DCs.20 23 Numerous studies have demonstrated the antitumor effects of GM-CSF, including promoting the maturation of DCs, inducing the differentiation of monocytes into M1 macrophages, and converting TAMs into M1 phenotypes.17 18 To study whether anti-S15×GM CSF is better than GM-CSF alone in reprogramming macrophages from immunosuppressive M2 phenotype to inflammatory M1 macrophages, we pretreated PBMCs with M-SCF for 5 days to obtain M2 macrophages, and then treated these macrophages with anti-S15×GM CSF and control antibodies. As shown in figure 4A, the dose-dependent effect of anti-S15×GM CSF on the reprogramming of macrophages was further validated by FACS assay. And anti-S15×GM CSF markedly upregulated the mRNA expression of inflammation biomarker IL-6 and downregulated the anti-inflammation biomarkers CD163, IL-10 and Arg-1 (figure 4B). These findings suggest that anti-S15×GM CSF may serve as a promising agent in antitumor immunotherapy.

Anti-S15×GM CSF reprograms macrophages to enhance their antigen uptake, processing and surface presentation. (A,B,D) CD14 cells were isolated from human PBMCs, induced with M-CSF for 5 days, and then stimulated with Anti-S15×hGM CSF or equal quantity of Isotype control antibody for 3 days. (A) CD80 or CD163 expression was determined by flow cytometry (n=3). (B) Extract total RNA and gene expression was determined by qPCR (n=4–6). (C) BM–derived macrophages from C57BL/6 mice were incubated with M-CSF for 5 days, and then stimulated with anti-S15×mGM CSF or equal quantity of isotype control antibody for 3 days. From left to right, they represent uptake antigen, processing antigen, antigen presentation MHC-I (H-2Kb) expression and MHC-II expression. Up panel, representative flow cytometry chart. Down panel, quantitative analysis (n=3). (D) From left to right, they represent uptake antigen, processing antigen, HLA-A2 expression and HLA-DR expression (n=3–4). Up panel, representative flow cytometry chart. Down panel, quantitative analysis (n=3). *p<0.05, **p<0.005, ***p<0.001, ****p<0.0001 versus the indicated. P values were determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test (A–D). BM, bone marrow; GM-CSFR, granulocyte-macrophage colony-stimulating factor receptor; hGM, human granulocyte-macrophage; IL, interleukin; MFI, mean fluorescence intensity; mGM, mouse granulocyte-macrophage; MHC, major histocompatibility complex; PBMC, peripheral blood mononuclear cell; qPCR, quantitative PCR.
Given that M1 macrophages display high capacity in antigen presentation,51 52 we next monitored whether anti-S15×GM CSF treatment can enhance macrophage antigen presentation. As antigen presentation by macrophages is generally separated into three phases: antigen uptake, antigen processing, and antigen presentation on cell surface,40 we monitored the three phases separately. First, mouse BMDMs treated with or without anti-S15×mGM CSF were incubated with FITC-Dextran. We found that the uptake of antigen by anti-S15×mGM-CSF-reprogrammed macrophages was significantly enhanced compared with non-treated macrophages (figure 4C). Second, to test whether macrophages can process the internalized antigen, macrophages treated with or without anti-S15×mGM CSF were incubated with a “self-quenching” molecule, DQ-ovalbumin (DQ-OVA).40 DQ-OVA has a high density of BODIPY fluorophore residues aligned along the OVA protein, which self-quench when the protein is intact but fluoresce when the protein is degraded. As shown in figure 4C, anti-S15×mGM CSF markedly increased the DQ-OVA fluorescence in macrophages, suggesting that the phagocytosed DQ-OVA was cleaved in anti-S15×mGM-CSF-reprogrammed macrophages. Finally, mouse macrophages treated with or without anti-S15×mGM CSF were pulsed with OVA or peptide OVA257–264 followed by 6-hour incubation in the presence or absence of anti-S15×mGM CSF. Cell surface antigen was detected by the peptide-specific antibody.40 Flow cytometry (figure 4C) showed that anti-S15×mGM CSF markedly increased the antigen presentation on the macrophage surface. In line with this, anti-S15×mGM CSF treatment markedly increased MHC-I/II expression on macrophage surface (figure 4C). Similarly, we observed enhanced antigen uptake and processing in human macrophages treated with anti-S15×hGM CSF (figure 4D). In summary, these results indicate that anti-S15×GM CSF is able to target reprogrammed macrophages to the M1 phenotype such as antigen up-taking, processing, and presentation.
Anti-S15×GM-CSF suppresses tumor growth
We next compared the half-life and biodistribution of anti-Siglec-15×mGM CSF with those of mGM-CSF in mice. In this experiment, equal numbers of anti-Siglec-15×mGM CSF and mGM-CSF molecules were intravenously injected into mice bearing with or without Siglec-15-expressing MC38 tumor. As shown in online supplemental figure S5A, cytokine half-life in mice was significantly extended in Anti-Siglec-15×mGM CSF (T1/2 =15.2 hours) compared with mGM-CSF alone (T1/2 =4.5 hours). This result is consistent with the previous report.53 In addition, different from mGM-CSF which was randomly distributed in various mouse organs and rapidly degraded following intravenous injection, anti-S15×mGM CSF enriched mGM-CSF in tumor tissue (figure 5B) and was readily detected on day 4 postinjection.

Anti-S15×GM CSF more effectively suppresses progression of mouse syngeneic tumor than Siglec-15 antibody. (A) Equal amount of mGM-CSF and anti-S15×mGM CSF was intravenously injected into C57BL/6 mice (n=3) and plasma was collected at indicated time points for determining the half-life by ELISA. (B–H) C57BL/6 mice were inoculated with mouse Siglec-15-expressing (mS15) MC38 tumor cells. (B) Postinoculation, mice were i.p. injected with anti-S15×mGM-CSF-Bio and equal amounts of mGM-CSF-Bio. After 4 days, tumors and various tissues were harvested and examined for distribution of GM-CSF by ELISA (n=5). (C) Schematic depiction of experimental design. Mice were i.p. injected with PBS, anti-S15, anti-S15×mGM CSF, and equal amount of mGM-CSF. Tumor growth was measured every 2 days (n=13). (D) Tumor growth curve in mice engrafted with MC38-mS15 tumor. (E) Representative tumor photographs (n=5). Scale bar=1 cm. (F) Average weights of the primary tumors collected from MC38 tumor-bearing mice after various treatments (n=5). (G) Mouse body weight (n=10–13). (H) The Kaplan-Meier survival curves of various treated groups (n=8). *p<0.05, ***p<0.001, ****p<0.0001. P values were determined by a two-way ANOVA with Sidak’s multiple comparisons (D) or one-way ANOVA with Tukey’s multiple comparisons test (B, F). ANOVA, analysis of variance; i.p., intraperitoneally; mGM-CSFR, mouse granulocyte-macrophage colony-stimulating factor receptor; ns, not significant; PBS, phosphate-buffered saline Siglec, sialic acid-binding immunoglobulin-like lectin.
We then assessed the effect of anti-S15×GM CSF on tumor progression in a mouse syngeneic model. MC38 is a mouse colon cancer that has been extensively used for immunotherapeutic intervention studies. A previous study did not observe any significant effects of anti-S15 antibody treatment on tumor growth,4 likely due to the low expression of Siglec-15 at the tumor site. To avoid this issue, we employed a mouse tumor model in which MC38 cells expressed mouse Siglec-15 (MC38-mS15).54 As depicted in figure 5C, mice were inoculated with MC38 cancer cells expressing mouse Siglec-15 on the right flank. After 1 week, mice were i.p. injected with anti-S15×mGM CSF or anti-S15 antibody (2 nM each, equal to 12 mg/kg for anti-S15, 6.38 mg/kg for mGM-CSF or 13.91 mg/kg for anti-S15×mGM CSF, respectively) every 2 days for a total of five injections. As shown in figure 5D–H, anti-S15×GM CSF displayed significantly higher efficacy in inhibiting tumor growth than anti-S15 antibody alone treatment.
To further test the efficacy of anti-Siglec-15 in different tumor models, we established mouse syngeneic tumor models using CT26, a mouse colon cancer cell line. The model has been reported to express Siglec-15 in tumor sites.4 55 As shown in online supplemental figure S5A, mice were inoculated with CT26 colon cancer cells on the right flank. One week later, anti-S15×mGM CSF or anti-S15 antibody (1.5 nM each, equal to 9 mg/kg for anti-S15 and 10.43 mg/kg for anti-S15×mGM CSF, respectively) was i.p. injected every 2 days for a total of five injections. As shown in online supplemental figure S5B–E, the effect of inhibiting tumor growth was significantly higher than that of anti-S15 antibody treatment alone.
In separated experiments, tumors were collected and analyzed by flow cytometry and ELISA. Cell analysis of TME indicated that anti-S15×mGM CSF treatment markedly increased the levels of CD45 cells, DCs and M1 macrophages in TME (figure 6B). Anti-S15×mGM CSF also showed better effect in promoting CD3 T cells particularly CD8 effector T cells in TME (figure 6C). In total CD45 cells, the percentage of DCs, M1 macrophages and CD8 T cells was markedly increased by treatment with anti-S15×mGM CSF but not anti-S15 antibody alone (figure 6D). Moreover, anti-S15×mGM CSF markedly increased the proinflammatory cytokines such as IL-6, TNF-α and decreased the anti-inflammatory cytokines such as TGF-β1 and IL-10 (figure 6E). These results collectively suggest that anti-S15×GM CSF suppresses tumor progression via reprogramming TME and activating antitumor immune cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Anti-S15×GM CSF is more effective than Siglec-15 antibody alone to suppress tumor progression in the mouse syngeneic tumor model. C57BL/6 mice were inoculated with mouse Siglec-15-expressing (mS15) MC38 tumor cells. (A) Schematic depiction of experimental design. (B–C) Flow cytometry analysis of CD45+ cells, DCs and ratio of M1/M2 macrophages (B), CD3+ cells, CD4+ cells and CD8+ T cells (C) in MC38-M15 subcutaneous tumor-bearing mice treated with PBS, anti-S15 or anti-S15×mGM CSF. (D) ELISA analysis of IL-10, TGF-β1, IL-6 and TNF-α levels in tumor sites from mice after various treatments. Data were presented as mean±SD (n=5). *p<0.05, **p<0.005, ***p<0.001 versus indicated. P values were determined by a one-way ANOVA with Tukey’s multiple comparisons test (B, C, E). ANOVA, analysis of variance; DC, dendritic cell; IL, interleukin; i.p., intraperitoneally; mGM-CSFR, mouse granulocyte-macrophage colony-stimulating factor receptor; PBS, phosphate-buffered saline; Siglec, sialic acid-binding immunoglobulin-like lectin.
Discussion
In the present study, we generate a chimera of anti-S15×GM CSF aiming to take advantage of both immune checkpoint Siglec-15 blockade and macrophage priming by GM-CSF in antitumor immunotherapy. Our results demonstrate that anti-S15×GM CSF has a superior function to anti-S15 antibody or GM-CSF alone in suppressing tumor progression. Mechanistically, anti-S15×GM CSF not only extends the half-life of GM-CSF but also specifically delivers GM-CSF to TAMs. The strong antitumor effect of anti-S15×GM CSF is likely dependent on its capacity to reprogram TME, resulting in a significant increase of DCs, inflammatory M1 macrophages, and CD8 T cells.
GM-CSF, a cytokine that can prime and activate immune cells, has been used in tumor immunotherapy.25 33 56 In particular, GM-CSF can induce DC maturation and reprogram M2 macrophages.17 18 20 23 However, a high concentration of GM-CSF in the serum will cause adverse side effects, including fever, nausea, diarrhea, etc. Therefore, selective delivery of GM-CSF to target cells and controlling the serum concentration of GM-CSF is a key to treating patients with tumor with GM-CSF. In the present study, we selected Siglec-15, which highly expresses on TAMs, as a target to specifically deliver GM-CSF to tumor sites, particularly TAMs. We thus designed a chimera of anti-S15×GM CSF, which well maintains the function of both Siglec-15 antibody and GM-CSF. As shown in the results, linking to Siglec-15 antibody not only markedly extended the half-life of GM-CSF, but also increased the TAM-targeting capacity of GM-CSF. These improvements strongly enhance the targeted efficacy of GM-CSF in the TME while reducing its adverse side effects on various organs and tissue, a challenge encountered in GM-CSF monotherapy.
Like PD-L1, immune checkpoint molecule Siglec-15 is a transmembrane protein expressed on many types of tumor cells and macrophages, especially M2 macrophages.57 Studies have shown that while Siglec-15 and PD-L1 exhibit a positive correlation at the overall level, they are rarely co-expressed on the same cell.58 Although its functional counter-receptor on T cells remains undefined, Siglec-15 has been shown to directly inhibit the cytotoxicity of CD8 T cells.4 7 8 Siglec-15 blockade using neutralizing antibodies has been recently tested in clinical trials by NextCure.10 Unfortunately, Siglec-15 antibody NC318 alone failed to achieve tumor remission response in phase II clinical trials.11 Given that tumors can coexpress different immune checkpoint molecules, Siglec-15 blockade may achieve better antitumor efficacy when combined with the antibodies against other immune checkpoint molecules. The reason we select Siglec-15 as the target is not only that TAMs highly express Siglec-15 and anti-S15 antibodies can efficiently deliver GM-CSF to TAMs, but also that Siglec-15 blockade itself can significantly promote antitumor T cell function. In other words, through generating anti-S15×GM CSF, we have achieved two goals with one molecule: (1) to block Siglec-15 by anti-S15 antibody to reduce the Siglec-15-mediated suppression of antitumor CD8 T cells, and (2) to target reprogram TAMs into proinflammatory M1-like macrophages by GM-CSF while minimizing the potential toxicity observed in GM-CSF monotherapy. In the present study, we did observe the effect of anti-S15×GM CSF on both activating CD8 T cells and promoting macrophage transformation and antigen presentation. Compared with anti-S15 antibody alone, anti-S15×GM CSF has a superior function in suppressing the progression of humanized Siglec-15-expressing MC38 tumors in mice (figure 5).
There are some limitations in our study that need to be addressed in future research. First, we only tested the effect of anti-S15×GM CSF in mouse models and a small number of cancer cell lines. Future studies should investigate the efficacy of this chimera in a broader range of cancer types and animal models. Second, the safety and toxicity of anti-S15×GM CSF should be further evaluated in preclinical trials. In addition, since we used human antibodies in the present study, potential antidrug antibodies generated in the mice in the long run and their effects on the mouse therapeutic response should be carefully examined. Through harnessing the function of both Siglec-15 blockade and GM-CSF, our studies have demonstrated that anti-S15×GM CSF is able to recapitulate the bioactivities mediated by Siglec-15 blockade and GM-CSF activation, resulting in potent antitumor synergistic efficacy. Taken together, the present study highlights anti-S15×GM CSF as a promising immunotherapeutic approach for cancer treatment.
Data availability statement
No data are available. Data is included within the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
The tissue microarrays (HNT961 and LUC1501) were obtained from Guilin Fanpu Biotech company (National Human Genetic Resources Sharing Service platform No. 2005DKA21300), under the approval by the Ethics Committee of Tong Xu First Hospital. Participants gave informed consent to participate in the study before taking part.
References
Footnotes
ZM, XH, SQ and QZ contributed equally.
Contributors KZ and MC designed experiments. ZM, XH, SQ and QZ performed experiments. SG and DZ analyzed data. QZ, JLuo and SG provided critical samples. HL, JLiu, WD and JLi contributed technical or material support. KZ and ZM wrote the paper. ZM, KZ and MC are the guarantors.
Funding This work was supported by grants from the Ministry of Science and Technology of China (2018YFA0507100), National Natural Science Foundation of China (82170692) and China Postdoctoral Science Foundation (2021M703595) and Jiangsu Funding Program for Excellent Postdoctoral Talent (2022ZB284).
Competing interests XH, HL, JLiu and WD, JLi and MC are employees of Biosion. These authors receive compensations and stocks of Biosion. The remaining authors declare no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.