Article Text

Abstract
Background Major histocompatibility complex class I-related protein A and B (MICA/B) are ligands for the natural killer group 2 member D (NKG2D) receptor and are broadly expressed on tumor cells but minimally on normal tissues. When cytotoxic NKG2D-expressing immune cells engage MICA/B, the ligand-expressing cells are targeted for lysis. Cancer cells can evade NKG2D-mediated destruction by shedding MICA/B from their cell surface via proteases present in the tumor microenvironment. CLN-619 is a humanized IgG1 monoclonal antibody (mAb) which binds MICA/B and inhibits shedding resulting in accumulation of MICA/B on the tumor cell surface. CLN-619 may thereby have therapeutic effects in a broad range of malignancies by re-establishing the MICA/B-NKG2D axis to enable NKG2D-mediated, as well as Fc-gamma receptor-mediated, tumor cell lysis.
Methods CLN-619 was characterized for binding epitope and affinity, effects on surface and soluble levels of MICA/B, and in vitro tumor cell killing. In mouse models, the mAb was tested for tumor growth inhibition. The contribution of the Fc-gamma (Fcγ) 1 domain to CLN-619 activity was also assessed.
Results CLN-619 bound with high affinity to the alpha-3 domain of MICA/B without encumbering the interaction with NKG2D on natural killer cells. CLN-619 increased the level of cell surface expression of MICA/B and concomitantly decreased the levels of soluble MICA/B in cell culture assays. Treatment of cancer cell lines with CLN-619 induced antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis. CLN-619 resulted in potent inhibition of tumor growth in multiple xenograft models and increased survival of mice in a disseminated cancer model.
Conclusions CLN-619 inhibited the shedding of MICA/B to effectively restore cytotoxic signaling pathways in immune cells. Potent antitumor activity of CLN-619 as a monotherapy was observed in several preclinical models. Activity of CLN-619 required a functional Fcγ1 domain, suggesting the requirement of simultaneous engagement of NKG2D and cluster of differentiation 16A (CD16A) on immune cells for optimal cytotoxicity. The preclinical data reported here support the assessment of CLN-619 in patients with cancer.
- Monoclonal antibody
- Natural killer - NK
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
In the clinic, efforts are ongoing to leverage natural killer (NK) cells for immunotherapy including cell therapies that engage the natural killer group 2 member D (NKG2D) pathway.
Challenges of targeting major histocompatibility complex class I-related protein A and B (MICA/B) include the large number of distinct MICA/B alleles in the population, shedding of the ligands in the tumor microenvironment, and the potential need to engage additional receptors beyond NKG2D to obtain optimal immune cell activation.
Preclinical data on monoclonal antibodies (mAbs) designed to prevent shedding of MICA/B and restore the interaction between MICA/B and the NKG2D receptor have been described.36
WHAT THIS STUDY ADDS
We describe discovery efforts for CLN-619, a clinical stage humanized IgG1 mAb, that has broad reactivity across the majority of MICA/B alleles, prevents shedding and thus increases cell surface MICA/B, and simultaneously engages the NK cell activating receptors NKG2D and cluster of differentiation 16A (CD16A).
Preclinical studies demonstrate high-affinity binding of CLN-619 to the alpha-3 domain of MICA and stabilization of cell surface MICA/B resulting in potent NK cell-mediated killing of MICA/B-expressing target cells. In vivo studies in human MICA/B-expressing xenograft mouse models show single-agent antitumor activity of CLN-619.
CLN-619 was the first anti-MICA/B targeted mAb to enter human clinical trials.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The preclinical data described here, including demonstration of potent single-agent efficacy at low doses in multiple human xenograft models, supports further evaluation of CLN-619 in an ongoing clinical trial in patients with solid tumors (NCT05117476).
Introduction
The natural killer group 2 member D (NKG2D) receptor is expressed on natural killer (NK) cells as well as on certain T-cell populations, including cytotoxic CD8+T cells, gamma/delta (γδ) T cells and natural killer T (NKT) cells.1 2 NKG2D recognizes eight glycoprotein ligands, including major histocompatibility complex class I-related protein A and B (MICA and MICB) and UL16 binding proteins 1–6.1 3 NKG2D ligands are upregulated in response to cellular stress including viral infection, carcinogenesis and genotoxic damage. On engagement of one or more ligands, NKG2D triggers cytotoxic lysis of the ligand-expressing cell, thereby eliminating damaged cells.4–6 Of the NKG2D ligands, the MICA/B ligands are most broadly expressed in hematologic and solid tumor malignancies with minimal normal tissue expression, making them attractive pan-cancer targets.7 Preclinical data validates the role of the MICA/B-NKG2D axis in immune surveillance, as overexpression of MICA in human tumor xenograft models results in delayed tumor growth and increased survival of mice,8 whereas NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy.5
While all NKG2D ligands contain alpha-1 and alpha-2 domains which functionally bind a dimeric NKG2D receptor, only MICA and MICB include an additional alpha-3 domain, which contains multiple protease cleavage sites.9–12 Cancer cells can escape NKG2D-mediated lysis by shedding MICA/B from the cell surface via proteases released in the tumor microenvironment (TME).13 14 Soluble MICA (sMICA) is a negative prognostic marker in a variety of cancers.15 In contrast, expression of MICA on the surface of cancer cells, as observed in tumor biopsies, is a positive prognostic factor for improved overall survival for several cancers.15
Given the compelling biology of the NKG2D-MICA/B axis, diverse efforts are underway to leverage this pathway for cancer therapy. Approaches targeting NKG2D ligand-expressing cancer cells include genetically engineered T or NK cells, NK cell engagers, antibody drug conjugates, vaccines and monoclonal antibodies (mAbs).7 In particular, novel therapies that engage NK cells through more than one activating receptor, for example, NKG2D plus CD16A, may provide an alternative to T-cell therapies.
Here, we describe CLN-619, a humanized IgG1 (hIgG1) mAb that targets MICA/B and is currently in early clinical development (NCT05117476). CLN-619 was designed to prevent shedding of MICA/B thereby increasing surface expression of these ligands on tumor cells to re-establish the MICA/B-NKG2D axis and enable immune-mediated tumor cell lysis.
Materials and methods
Generation of CLN-619
A repetitive immunizations multiple sites immunization protocol was carried out in Swiss James Lambert mice. The full-length extracellular domain (ECD) of the most common allelic variant, MICA*008, was used as the antigen.16 The lead candidate was humanized using in silico modeling.
Octet binding assessment of monovalent affinity of CLN-619 to MICA and MICB
CLN-619 at 10 micrograms per milliliter (μg/mL) was captured on anti-human IgG Fc capture biosensors. Sensors were then incubated with purified MICA ECD proteins. A fitting algorithm (ForteBio), which assumed 1:1 binding, was used to calculate association (ka), dissociation (kd), and equilibrium dissociation (KD) constants.
ELISA assessment of CLN-619 binding to MICA/B allelic variants
Plates coated with monomeric histidine (His)-tagged MICA/B were incubated with a serial dilution of CLN-619. Horseradish peroxidase (HRP) conjugated mouse anti-human IgG Fc was used for detection.
Luminex assessment of CLN-619 binding to MICA
Binding of CLN-619 (10 µg/mL) and 6D4, a positive control antibody, to 28 allelic variants of MICA was assessed by Luminex.17 Goat anti-mouse secondary antibody was used to detect 6D4. Background mean fluorescence intensity (MFI) was subtracted from the raw MFI and duplicates were averaged.
X-ray crystallography of MICA-CLN-619
X-ray diffraction data were collected from crystals of fragment antigen binding (Fab) CLN-619:MICA using optimized cryogenic conditions. The structure of the complex of human Fab antibody fragment CLN-619 with MICA alpha-3 domain (Fab CLN-619:MICA) was at 2.12 angstrom (Å) resolution.
Flow cytometry assessment of surface MICA/B levels following CLN-619 treatment
Cells were incubated with serially diluted CLN-619, CLN-619 DANA or hIgG1 control antibody for 24 hours. CLN-619 DANA contains two mutations in the Fcγ domain, D265A and N297A, previously shown to abolish Fc-gamma receptor (FcγR) binding of hIgG1 antibodies.18 Cells were stained with live/dead viability dye. MICA/B was detected with 6D4-phycoerthrin (PE) antibody, which is non-competitive with CLN-619. Mouse IgG2a-PE isotype antibody was used as a control. Surface MICA/B levels were quantified with Quantibrite beads. Dose response curves were fit using a four-parameter logistic regression model.
Assessment of soluble MICA/B levels following CLN-619 treatment
Cells were treated with serially diluted CLN-619 or hIgG1 in duplicate for 24 hours and supernatant was collected for analysis by ELISA. Antibodies used in the ELISA were non-competitive with CLN-619. Plates were coated with BAMO1 capture antibody. A standard curve of recombinant human MICA*001 ECD was used. Biotinylated detection antibody 10E9.H6 was added followed by streptavidin-HRP. Half-maximal inhibitory concentration (IC50) values were calculated using a four-parameter non-linear fit model.
NKG2D and FcyRIIIa/CD16A reporter assays
Induction of CD16A or NKG2D signaling by CLN-619 was measured using a reporter assay. Jurkat cells expressing high (V) or low affinity (F) FcyRIIIa/CD16A (Promega), NKG2D, or NKG2D with a truncated high affinity CD16A (V176) were engineered to express nuclear factor of activated T cells (NFAT)-luciferase reporter construct. HCT-116, HCC1534, MICA expressing Chinese hamster ovary (CHO) or CHO control cells were treated with a serial dilution of CLN-619, CLN-619 DANA, or hIgG1 control antibody. Engineered Jurkat cells were added in an effector:target (E:T) ratio of 10:1 for 6 hours with human cell lines or E:T ratio of 25:1 for 24 hours with CHO cell lines. Half-maximal effective concentration(EC50) values were calculated using the four-parameter non-linear fit model.
Primary NK cell killing assay
NK cell-mediated cytotoxicity was assessed via co-culture of purified naïve human primary NK cells and MICA/B-expressing HCC1534 tumor cells at a 40:1 E:T ratio in a xCELLigence system for 72 hours. NK cells were isolated through negative selection from frozen peripheral blood mononuclear cells (PBMC) stocks from healthy human donors and checked for purity (online supplemental figure 1). EC50 values were calculated using a four-parameter non-linear fit model.
Supplemental material
Primary cell antibody-dependent cellular phagocytosis assay
Cluster of differentiation (CD) CD14+ monocytes were collected from two healthy PBMC donors by magnetic positive selection. Monocytes were differentiated into macrophages by exposure to macrophage colony-stimulating factor supplemented media for 6 days. Differentiation was confirmed microscopically. eFluor 670-labeled target cells and effector cells were plated at an E:T ratio of 0.5:1 and incubated for 2 hours with serially diluted CLN-619, hIgG1 or cetuximab control antibodies at the highest dose. After incubation, macrophages were successively incubated with viability dye, blocking buffer, and CD64-Brilliant Violet 510 (BV510) antibody. Antibody-dependent cellular phagocytosis (ADCP) was determined via flow cytometry by gating target cell fluorescence (eF670+) within CD64+ cells.
Flow cytometry assessment of MICA binding to NKG2D on NK cells
NK cells were purified from healthy donor PBMCs by negative selection. Serially diluted CLN-619, CLN-619 DANA or hIgG1 antibody were preincubated with His6-tagged MICA ECD at a ratio of 10:1. Antibody-MICA complexes were added to the NK cells in the presence or absence of anti-NKG2D antibody. MICA bound to NK cells was detected by anti-His6 antibody.
Biacore assessment of CLN-619 to Fc gamma receptors
Recombinant His6-tagged FcγRs were immobilized onto an anti-His6 coated CM5 chip. CLN-619 or CLN-619 DANA were flowed over the biosensor. ka, kd and KD were calculated at steady state for all FcRs except CD64, which was evaluated at 1:1 affinity.
Flow cytometry assessment of NKG2D on murine and human NK cells
Peripheral blood cells were collected from mice and analyzed by flow cytometry.
In vivo efficacy studies
Female BALB/c SCID mice 6–9 weeks of age (N=10/group) were inoculated subcutaneously with 5×106 PLC/PRF/5 cells or 1×107 HCC1534 cells. The PLC/PRF/5 model was dosed by intraperitoneal (i.p.) injection with either vehicle (phosphate buffered saline) or CLN-619 (0.3, 3.0 milligrams per kilogram (mg/kg)) three times a week for a total of 16 doses. The HCC1534 model was treated with hIgG1 (10 mg/kg), CLN-619 (0.03, 0.3, 3.0, 10 mg/kg), or CLN-619 DANA (0.03, 0.3, 3.0 mg/kg). Intraperitoneal dosing began on the same day as tumor cell implantation and continued twice weekly for 4 weeks. Tumor size and body weight were measured twice a week. PLC/PRF/5 and HCC1534 studies ended on days 39 and 35, respectively. Statistical analysis was performed using two-way analysis of variance (ANOVA) with multiple comparisons. Animal welfare for these studies complies with the US Department of Agriculture’s Animal Welfare Act (9 CFR Parts 1, 2 and 3) as applicable and was covered by the Institutional Animal Care and Use Committee (IACUC) (protocol CBSD-ACUP-001).
Immunodeficient hIL-15-NOG female mice aged 8–12 weeks were i.p. injected with 5×106 PBMC from a normal human donor. On day 8, mice were subcutaneously engrafted with 2×107 A2058 cells and treated with CLN-619, CLN-619 DANA or hIgG control (dosed i.p. twice weekly) (N=15/group). Two-way ANOVA multiple comparisons were completed on day 37, the last day all control animals were on study.
HCT-116 luciferase-tagged cells were inoculated by intravenous injection in 6–8-week-old female BALB/c SCID mice. Treatment began on day 1 with hIgG1 or CLN-619 dosed at 10 mg/kg administered twice a week for 6 weeks by i.p. injection. Bioluminescent imaging was performed twice weekly to monitor disease progression. Survival analysis was performed on day 104, the defined study endpoint. Statistical analysis was performed using a log-rank test. All procedures were carried out under the institutional guidelines of Translational Drug Development IACUC (Protocol #22011).
Pharmacokinetics
BALB/c SCID mice were dosed i.p. with 1, 3 or 10 mg/kg of CLN-619, and blood samples were collected by mandibular bleeds post-dosing. A total of three or four mice were assessed at each time point in each dose group. For analysis, ELISA plates were coated with recombinant MICA*001 ECD. A standard curve of CLN-619 or serum samples diluted 1:250 was added to the plates. Mouse anti-human HRP secondary detection antibody was added to the plates. Non-compartmental data analysis was performed.
Results
CLN-619 binds to the alpha-3 domain of MICA/B
CLN-619 was derived from a parental murine antibody that was selected for high affinity binding to the alpha-3 domain of MICA and the ability to augment cell surface expression of MICA/B. The variable domains of the parental antibody were humanized and introduced into a human IgG1 backbone to generate CLN-619.
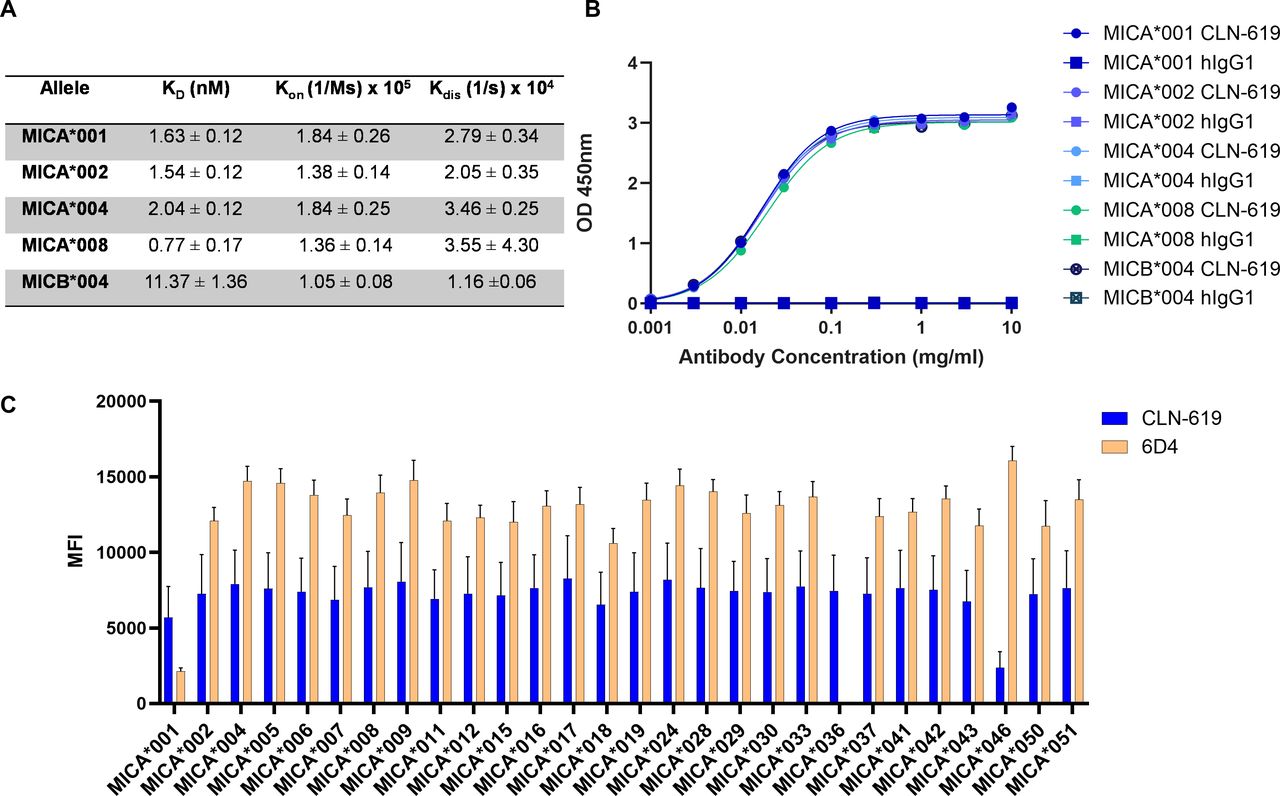
The MICA/B genes are highly polymorphic with nearly 150 alleles of MICA and 50 alleles of MICB present in the human population.16 The canonical MICA*001 and MICB*004 alleles are 83% identical in overall protein sequence and 91% identical in the alpha-3 domain (Thr204 to Ser297).19 Given the highly polymorphic nature of the proteins, it was critical to confirm broad reactivity of CLN-619. The monovalent affinity of CLN-619 for the ECD of several common human MICA alleles (MICA*001, *002, *004, and *008) and the MICB*004 was measured by Octet. KD values for CLN-619 binding to recombinant MICA allelic variants ranged from 0.77 to 2.04 nanomolar (nM), that is, all within threefold (figure 1A). The KD value of CLN-619 binding to the MICB*004 was 11.37 nM, that is, approximately 5-fold to 15-fold lower affinity than for the MICA alleles. ELISA experiments independently confirmed binding of CLN-619 to the representative allelic variants (figure 1B). The broad reactivity of CLN-619 was further confirmed in a Luminex-based assay assessing 28 recombinant human MICA ECD proteins representing the most prevalent alleles (figure 1C). CLN-619 showed similar binding to all alleles, except for MICA*046, a rare allele present in less than 0.01% of the population, where binding was reduced (online supplemental table 1). Overall, CLN-619 exhibited broad reactivity across all MICA alleles tested, which represent 89% of the population.

Binding characteristics of CLN-619. CLN-619 binding to representative MICA allelic variants and the canonical MICB allelic variant as measured (A) by Octet (N=3; N refers to biological replicates throughout) and (B) by ELISA (N=2). CLN-619 had similar levels of binding to the various proteins; therefore, not all symbols are visible. (C) CLN-619 binding to 28 of the most common MICA allelic variants as measured by Luminex (N=2). 6D4 was used as a positive control. Error bars represent SEM. ka, association constant; kd, dissociation constant; KD, equilibrium dissociation constant; OD, optical density; MFI, mean fluorescence intensity; MICA/B, major histocompatibility complex class I-related protein A/B; SEM, standard error of the mean.
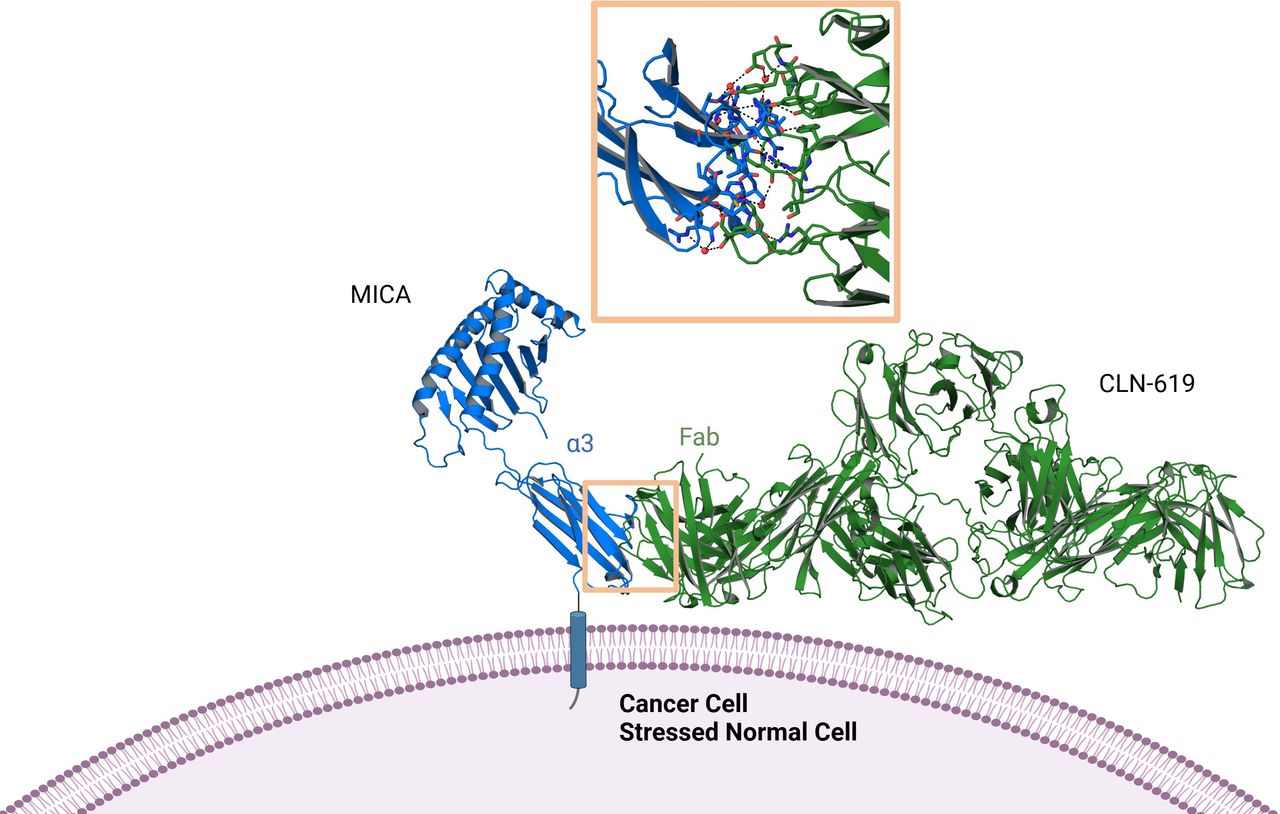
An X-ray crystallographic structural analysis was carried out to elucidate the interaction between CLN-619 and the alpha-3 domain of MICA. A co-crystal structure of the CLN-619 Fab fragment and the MICA alpha-3 domain of MICA*001 was obtained at a resolution of 2.12 Å. CLN-619 recognized a discontinuous epitope on the alpha-3 domain interacting with a total of 19 amino acid residues that cover approximately 20% of the alpha-3 domain. Notably, several of the known cleavage sites within the alpha-3 domain of MICA/B overlap with the epitope recognized by CLN-619.12 The CLN-619 epitope is highly conserved across the most common MICA allelic variants, with amino acid changes observed in only 4 of the 19 interacting residues, with most of these changes being relatively conservative (online supplemental table 2). Importantly, CLN-619 bound the alpha-3 domain of MICA in a region and at an angle that should not sterically interfere with binding of the alpha-1 and alpha-2 domains to the NKG2D dimer (figure 2).

CLN-619 co-crystal structure with MICA. X-ray crystal structures of the Fab fragment of CLN-619 in complex with the MICA*001 alpha-3 domain at 2.12 Å resolution. The orange box magnifies the binding interface of CLN-619, which is a discontinuous epitope comprising 19 amino acid residues exclusively in the alpha-3 domain of MICA. Å, angstrom; Fab, fragment antigen binding; MICA, major histocompatibility complex class I-related protein A.
Supporting the crystallographic analysis, CLN-619 did not hinder the NKG2D-MICA interaction in a flow-based assay. Interestingly, when MICA was complexed with CLN-619, increased NK cell binding was observed compared with MICA alone, likely reflecting simultaneous engagement of CLN-619 with both NKG2D and CD16A on NK cells (online supplemental figure 2). When an anti-NKG2D antibody was included in the experiment, MICA binding to primary NK cells was significantly reduced.
CLN-619 augments cell surface expression of MICA/B on tumor cells
Based on the structural data, we hypothesized that CLN-619 binding to MICA/B in the alpha-3 domain may prevent access of proteases to MICA/B ligands thereby resulting in reduced shedding and increased cell surface expression. We assessed the ability of CLN-619 to modulate levels of cell surface and soluble MICA/B (sMICA/B) in cell-based assays using human cancer cell lines HCC1534 (lung), PLC/PRF/5 (liver), and HCT-116 (colorectal), which express approximately 58,000, 17,000 and 10,000 copies of MICA/B on their surface, respectively, as measured by flow cytometry. Following treatment with CLN-619, cell surface levels of MICA/B were assessed by flow cytometry and sMICA/B levels present in culture media were measured by ELISA. CLN-619, but not control hIgG1, augmented the surface expression of MICA/B (figure 3A, online supplemental figure 3) and concomitantly decreased the levels of sMICA/B in a dose-dependent fashion (figure 3B). EC50 values for increased MICA/B surface expression were 54.3, 69.4 and 50.7 nanograms/milliliter (ng/mL) for HCC1534, PLC/PRF/5 and HCT-116 cell lines, respectively. IC50 values for inhibition of sMICA/B release into the cell culture medium were 107, 162 and 78.9 ng/mL CLN-619, respectively, for the three cell lines. Notably, the similar EC50 values for enhancement of MICA/B on the cell surface by CLN-619 and IC50 values for reduction of sMICA/B in the supernatants support a causal relationship.

CLN-619 modulates cell surface MICA/B through inhibition of shedding, while also engaging CD16A to enhance NKG2D signaling. (A) Cell surface levels of MICA/B were measured by flow cytometry. (B) Soluble MICA/B in the cell supernatant was measured by ELISA (N=3 or N=2). Geometric mean EC50 values and 95% CIs are reported. (C) MICA-expressing target cells or control cells treated with serially titrated CLN-619 or CLN-619 DANA were co-cultured with NFAT-luciferase Jurkat cells expressing NKG2D or NKG2D and truncated CD16A with an E:T of 25:1 for 24 hours (N=3). T-test values from area under the curves are reported for each reporter line. Control CHO and isotype (500,000 pg/mL) treatment responses were close to baseline and cannot be visualized. E:T control background subtracted. Error bars represent SEM. EC50, half-maximal effective concentration; CHO, Chinese hamster ovary; E:T, effector:target; IC50, half-maximal inhibitory concentration; MICA/B, major histocompatibility complex class I-related protein A/B; NFAT, nuclear factor of activated T cells; NKG2D, natural killer group 2 member D; NS, not significant; RLU, relative luminesence units; sMICA/B, soluble MICA/B.
CLN-619 induces NKG2D signaling and promotes cooperativity between the NKG2D and CD16A receptors
Given that CLN-619 stabilized MICA/B on the cell surface, we investigated whether this potentiates stimulation of NKG2D signaling. Jurkat T cell lines with luciferase gene expression driven under control of the NFAT transcription factor in response to human NKG2D activation were used as reporter effector cells, and a CHO cell line engineered to stably express human MICA or a control CHO line were used as target cells. In the reporter system, CLN-619 was shown to stimulate NFAT signaling through NKG2D engagement in a dose-dependent manner (figure 3C).
An Fc-silenced version of CLN-619, referred to as CLN-619 DANA, was generated to evaluate the contribution of FcγR engagement to CLN-619-mediated activity. CLN-619 DANA contains two mutations in the Fcγ domain, D265A and N297A, previously shown to abolish FcγR binding of hIgG1 antibodies.18 While the DANA mutations eliminated FcγR binding by CLN-619, the ability to bind MICA/B and modulate cell surface levels was retained (online supplemental table 3 and figure 4). In the reporter system, CLN-619 DANA was able to stimulate NKG2D signaling relatively similar to CLN-619 (p=not significant), demonstrating that the Fcγ1 domain of CLN-619 is dispensable for CLN-619-mediated NKG2D activation (figure 3C). When a truncated signaling-null version of CD16A was stably co-expressed in the Jurkat-NKG2D system, significantly enhanced NKG2D signaling (four-fold; p=0.0003) was observed on treatment with CLN-619 at the top concentration compared with CLN-619 DANA. This data is consistent with the enhanced MICA binding to NKG2D observed with CLN-619 compared with CLN-619 DANA (online supplemental figure 2), suggesting CD16A binding can further potentiate CLN-619-mediated NKG2D signaling.
CLN-619 potentiates NK cell-mediated cytokine production and target cell lysis
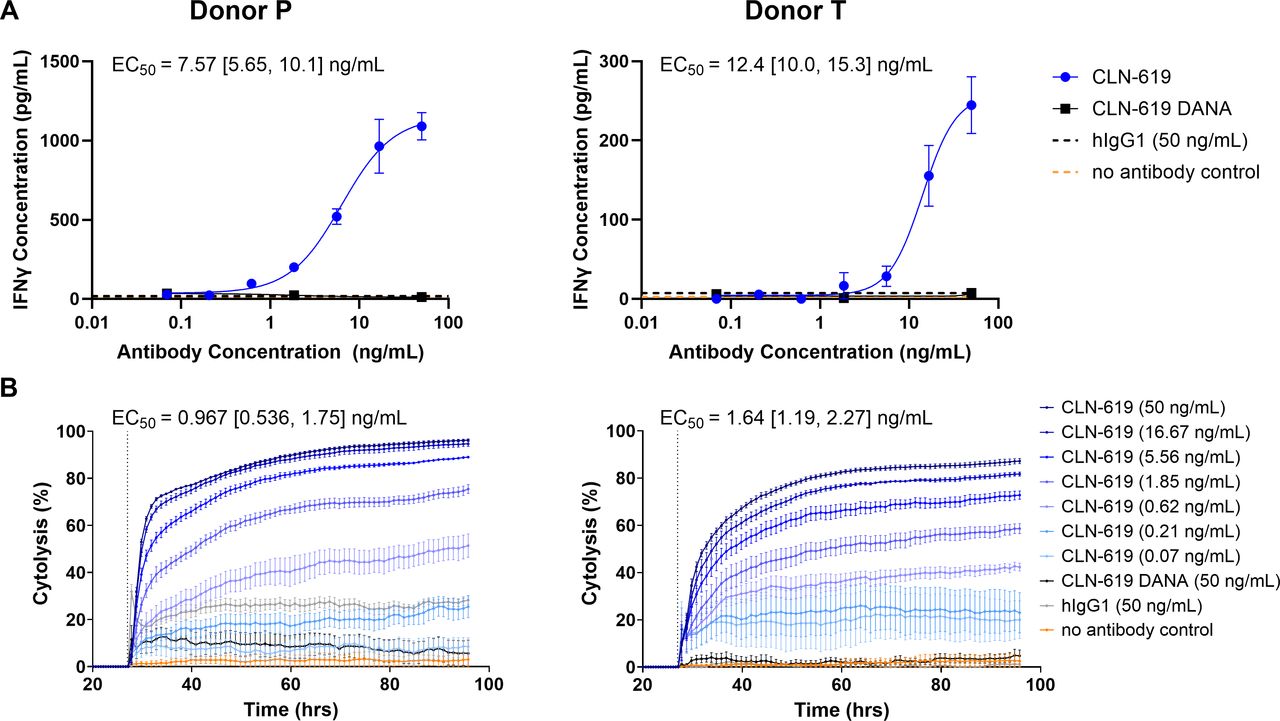
The ability of CLN-619 to stabilize MICA/B on the tumor cell surface and augment NKG2D signaling suggested that CLN-619 may be capable of potentiating NK cell cytokine release and target cell lysis. This was evaluated in a co-culture assay using primary NK cells derived from two healthy PBMC donors (online supplemental figure 1). In the presence of HCC1534 target cells, CLN-619 induced a dose-dependent release of interferon (IFN)-γ, a marker of NK cell activation, with EC50 values of 7.57–12.4 ng/mL (figure 4A), and target cell lysis with EC50 values of 0.967–1.64 ng/mL (figure 4B). When evaluated in the primary NK cell co-culture assay, CLN-619 DANA was not capable of eliciting significant IFN-γ release or target cell lysis (figure 4A,B). These results demonstrate the requirement of an intact Fcγ1 domain for CLN-619-mediated NK cell activation and target cell lysis.

CLN-619 mediates NK cell activation and killing of MICA/B-expressing target cells. (A) An ELISA (R&D Systems) was used to measure IFN-γ in the supernatants collected from a 72 hour co-culture killing assay with NK cells and MICA/B expressing target cells (E:T 40:1) in the presence of serially titrated antibody in two donors (N=2). Representative curves are shown. (B) xCELLigence was used to measure MICA/B-expressing target cell death from experiment described in (A). EC50 values were calculated for the last time point collected for each of the three runs. Representative curves are shown. For both panels, geometric mean EC50 and 95% CI values are reported. Error bars represent SEM. EC50, half-maximal effective concentration; E:T, effector:target; IFN, interferon; MICA/B, major histocompatibility complex class I-related protein A/B; NK, natural killer.
CLN-619 induces antibody-dependent cellular cytotoxicity and ADCP of MICA/B-expressing cells
Given that CLN-619 is an hIgG1 antibody with a functional Fc domain, we evaluated whether CLN-619 was capable of mediating various Fc-mediated functional activities, such as antibody-dependent cellular cytotoxicity (ADCC), ADCP and complement-dependent cytotoxicity (CDC). Engagement of CD16A by CLN-619 was investigated in a cell-based reporter assay as a surrogate for evaluating ADCC activity. Jurkat cell lines with luciferase gene expression driven under the control of NFAT in response to human CD16A activation were used as reporter effector cells. The Jurkat cell lines, expressing either the higher affinity V158 variant of CD16A and/or the lower affinity F158 variant,20 were co-cultured with MICA/B expressing target cells, HCC1534 and HCT-116, in the presence of CLN-619. Stronger CD16A signaling was observed on co-culture with HCC1534, the tumor cell line expressing higher levels of MICA/B (figure 5). The EC50 values ranged from 8 to 81 ng/mL with CLN-619 treatment, with the signal consistently lower for the F variant compared with the V variant of CD16A. CLN-619 DANA was inactive in the reporter assays. Overall, EC50 values for CD16A activation by CLN-619 in the reporter cell lines correlated with expression levels of MICA/B on target cells, suggesting that CLN-619 can mediate ADCC in a target-dependent manner. The activity was dependent on the presence of MICA as demonstrated by the lack of activity in the control CHO cells described above (online supplemental figure 5).

CLN-619 induces ADCC. MICA/B-expressing target cells treated with serially titrated antibody (CLN-619 or CLN-619 DANA) were co-cultured with NFAT-luciferase Jurkat cells expressing the human FcγRIIIa (CD16A) high affinity (V158) variant or low affinity (F158) variant at an E:T ratio of 10:1. Luminescence activity was quantified after 6 hours (N=3). Geometric mean EC50 values and 95% CIs for CLN-619 are reported. E:T control background subtracted. Error bars represent SEM. ADCC, antibody-dependent cellular cytotoxicity; EC50, half-maximal effective concentration; E:T, effector:target; F, phenylalanine; MICA/B, major histocompatibility complex class I-related protein A/B; NFAT, nuclear factor of activated T cells; RLU, relative luminescence units, V, valine.
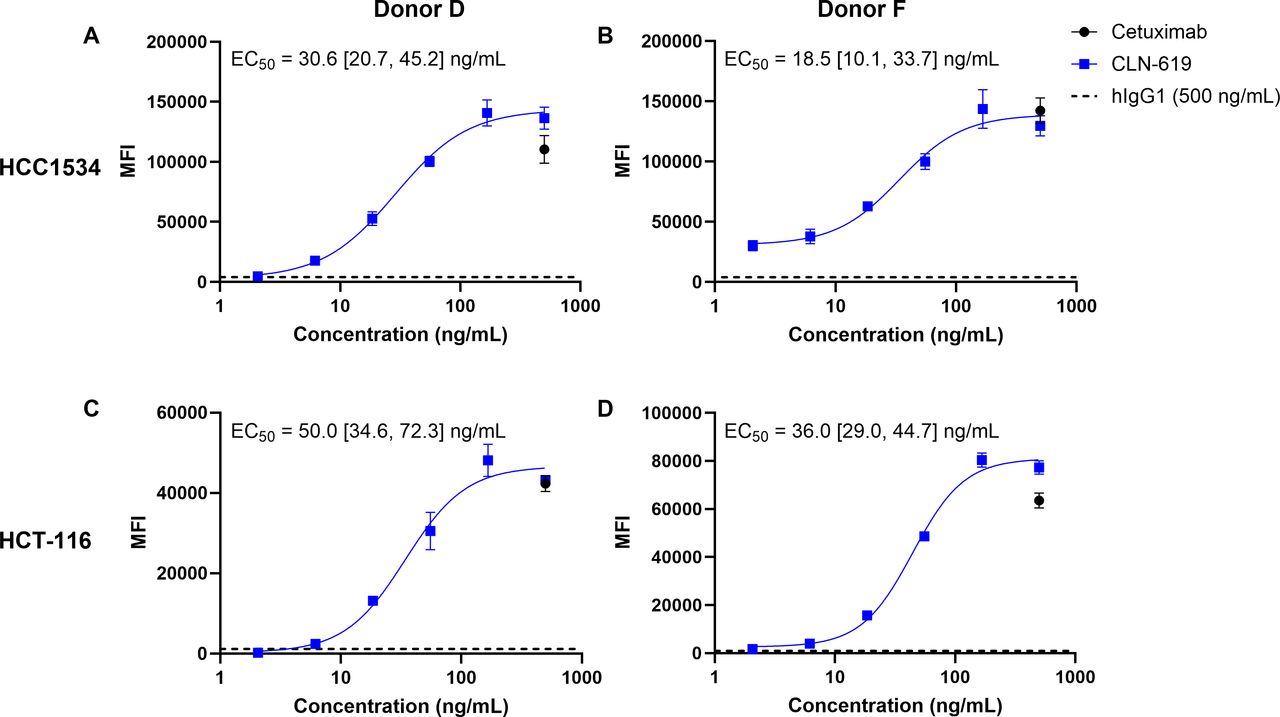
Given the ability of CLN-619 to also bind FcγRIIA/CD32A (online supplemental table 3), the FcγR receptor primarily responsible for ADCP, CLN-619 was next evaluated for its ability to elicit macrophage-mediated phagocytosis.21 Human peripheral monocytes that had been polarized to macrophages of the M1 phenotype were co-cultured with fluorescently-labeled HCC1534 or HCT-116 tumor lines. Phagocytosis in response to CLN-619 was measured by flow cytometry (online supplemental figure 6). The anti-epidermal growth factor receptor (EGFR) mAb cetuximab was included as a positive control, as both cell lines were confirmed to express EGFR. Treatment with CLN-619 resulted in dose-dependent phagocytosis of labeled tumor cells similar to cetuximab, while minimal phagocytosis was observed with an isotype control antibody (figure 6, online supplemental figure 6).

CLN-619 promotes ADCP. Macrophage-mediated phagocytosis of fluorescently labeled target cells was measured by flow cytometry. Representative curves for treatment of HCC1534 and HCT-116 cells with CLN-619 in two donors (N=3). Cetuximab and isotype control antibody treatment were tested at the highest concentration. Geometric mean EC50 values and 95% CIs are reported. Error bars represent SEM. E:T control background subtracted. ADCP, antibody-depedent cellular phagocytosis; EC50, half-maximal effective concentration; E:T, effector:target; MFI, mean fluorescence intensity.
While CLN-619 was capable of mediating ADCP and ADCC, no CDC activity was observed in response to CLN-619 treatment (data not shown).
Antitumor activity of CLN-619
The effect of CLN-619 on tumor growth in vivo was assessed with human tumor xenografts expressing MICA/B in immunodeficient BALB/c SCID mice. NK cells were confirmed to express high levels of NKG2D in this strain (online supplemental figure 7A). Exposure of CLN-619 following i.p. administration in BALB/c SCID mice was dose-proportional, and the serum half-life was in line with that of a typical human IgG1 antibody dosed in mice (online supplemental figure 8).
In the PLC/PRF/5 liver tumor xenograft model, i.p. administered CLN-619 at either 0.3 mg/kg or 3 mg/kg led to complete inhibition of tumor growth (p<0.0001) compared with the control group (figure 7A). In the HCC1534 lung xenograft model, i.p. administered CLN-619 at dose levels ranging from 0.3 to 10 mg/kg demonstrated statistically significant (p<0.0001) dose-dependent antitumor activity compared with treatment with isotype control, with tumor growth inhibition ranging from 45% to 87%. No antitumor activity was observed with CLN-619 DANA at any dose level (figure 7B). This data suggests that the intact Fcγ1 domain of CLN-619 is indispensable for antitumor activity in vivo, consistent with in vitro observations in NK cell co-culture assays.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CLN-619 treatment results in potent antitumor effects in several preclinical mouse models. (A) BALB/c SCID mice were inoculated with PLC/PRF/5 cells (N=10/group). Mice were treated 3× weekly i.p. with CLN-619 at two dose levels. (B) BALB/c SCID mice were inoculated with HCC1534 cells (N=10/group). Mice were treated twice weekly i.p. with CLN-619 or CLN-619 DANA. (C) huIL15-NOG mice were engrafted with human PBMC and inoculated with A2058 cells (N=15/group). Mice were treated twice weekly i.p. with CLN-619 or CLN-619 DANA. *p=0.002, **p<0.0001. For all three efficacy studies, statistics represent two-way ANOVAs with multiple comparisons. (D) BALB/c SCID mice were inoculated with HCT-116 luciferase-tagged human colon cancer cells (N=10/group). Mice were treated twice weekly i.p. with CLN-619 or hIgG1. Error bars represent SEM. ANOVA, analysis of variance; i.p., intraperitoneal; PBMC, peripheral blood mononuclear cells.
To confirm CLN-619 engagement of human NKG2D and CD16A pathways in vivo, efficacy was next evaluated in hu-IL15-NOG host mice engrafted with human PBMCs and implanted with A2058 melanoma cells. Notably, interleukin-15 mediates the survival and proliferation of NK cells as well as other immune cell types. Human NKG2D expression was confirmed in the donor (online supplemental figure 7B). CLN-619 administered at 3 mg/kg (i.p.) demonstrated statistically significant antitumor activity compared with hIgG1 control (p=0.002) and CLN-619 DANA (p<0.0001) (figure 7C).
The activity of CLN-619 in a metastatic setting was explored in a mouse model designed to mimic disseminated disease. HCT-116 human colorectal cancer cells expressing luciferase were inoculated intravenously allowing for tumor cell seeding to distal organs. Kaplan-Meier survival analysis demonstrated that CLN-619 treatment resulted in a statistically significant survival benefit as compared with hIgG1 (p=0.03) (figure 7D).
Discussion
CLN-619 is designed to overcome a potent immune escape mechanism exploited by cancer cells, whereby the mAb promotes accumulation of MICA/B on the tumor cell surface, thereby restoring the NKG2D-MICA/B axis and mediating ADCP and ADCC via its functional Fcγ1 domain to promote tumor cell lysis.
A challenge of generating MICA/B targeted therapies lies in the polymorphic nature of these NKG2D ligands. We show that CLN-619 has broad and high-affinity binding to MICA and MICB allelic variants. This is likely because CLN-619 binds to a highly conserved, discontinuous epitope in the alpha-3 domain. Importantly, CLN-619 does not interfere with receptor-ligand binding, preserving the NKG2D-MICA interaction. Of note, the binding epitope overlaps with known protease cleavage sites. Given the broad allelic reactivity of CLN-619, we expect that CLN-619 can stabilize and increase MICA/B expression in a heterogeneous population of patients with cancer.
The ability of CLN-619 to prevent MICA/B cleavage, presumably by steric hindrance of proteases, enables restoration of MICA/B expression on the tumor cell surface. Increased surface levels of MICA/B and decreased levels of sMICA/B were evident in multiple tumor cell lines in the presence of CLN-619. This reestablishment of MICA/B on the cell surface correlated with potent and dose-dependent release of IFN-γ and tumor cell lysis in primary co-culture assays of NK cells with cancer cell lines. We hypothesize the membrane proximal binding of CLN-619 to MICA (figure 2) enables robust immune synapse formation thereby promoting efficient tumor cell killing.
Our analyses revealed a multimodal mechanism of action of CLN-619. In addition to inhibition of MICA/B shedding and concomitant increased expression on the tumor cell surface, we observed CLN-619-induced ADCC and ADCP activity that was dependent on a wildtype hIgG1 backbone. An Fc-deficient version of CLN-619 was not active in the NK co-culture killing assay (figure 4B), despite retention of NKG2D engagement (online supplemental figure 2, figure 3C), and also lacked activity in vivo (figure 7B,C). We hypothesize that dual stimulation of CD16A and NKG2D receptors on NK cells may be required for the activity of CLN-619. This is supported by data from the reporter assay which demonstrated enhancement of NKG2D signaling when CD16A binding was enabled. This concept of dual receptor engagement driving NKG2D signaling is complimentary to published studies showing that NKG2D engagement can synergistically enhance ADCC4 22 and augment CD16A and natural killer cell p46-related protein (NKp46) activation of resting NK cells.23
CLN-619 showed compelling single-agent activity at low dose levels in multiple tumor xenograft models using tumor cell lines representing tumor types where the NKG2D pathway has been shown to be clinically relevant, such as hepatocellular carcinoma24 25 and non-small cell lung cancer.26 27 Human xenograft models were chosen because there is no mouse ortholog of MICA/B, while murine NKG2D on NK cells has been shown to recognize MICA/B on human tumor cells.28 29 The host mice used in the HCC1534 and PLC/PRF/5 studies have competent NK cells and macrophages but are devoid of T cells.30 Therefore, the observed antitumor efficacy is most likely attributable to target cell lysis by NK cells and/or phagocytosis by macrophages. In the A2058 humanized model, where the human immune system was reconstituted in a mouse strain that promotes NK cell engraftment, efficacy with CLN-619 was observed and was dependent on an intact Fc domain. This data further supports the potential role of NK cells in mediating CLN-619 activity, although the contribution of other immune cell types cannot be ruled out.
The NKG2D axis has been shown to play a role in CD8+T cell-mediated lysis of cancer cells and a costimulatory role in lowering the threshold for lysis by CD8+T cells.31 It can also drive CD8+T cell-mediated lysis of cancer cells following prior T-cell receptor activation in the absence of major histocompatibility complex expression.32 Future studies are needed to explore the potential contribution of CD8+, γδ T cells and NKT cells to the efficacy of CLN-619.
Therapies that potently engage NK cells may have advantages over therapies that exclusively engage T cells. NK cells do not require antigen priming for activation nor do they secrete high levels of cytokines that could trigger cytokine release syndrome, and they do not elicit graft-versus-host reactions. However, NK cells may require multiple activating stimuli to fully unleash their killing potential and to overcome inhibition by the many negative regulatory receptors expressed on NK cells. Moreover, NK cells are heterogeneously distributed in the TME, and different indications may have variable numbers of infiltrating NK cells.33–35 In several indications, a dysfunctional subset of NK cells was found to be enriched in the TME and correlated with unfavorable prognosis.33 A mechanism to improve NK cell performance may be the simultaneous engagement of synergistic NK cell-activating receptors, as described here for CLN-619. Additionally, a combination with therapies that enhance NK cell function, persistence and/or tumor infiltration could be beneficial and should be explored further.
Taken together, the data reported here support the clinical investigation of CLN-619 for the treatment of a broad range of tumor types. An ongoing phase I clinical trial of CLN-619 in cancer patients as a monotherapy and in combination with pembrolizumab is currently in progress (NCT05117476).
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
Rouken Bio performed the primary ADCP assays, NK co-culture killing assays and NKG2D signaling reporter assays. Proteros Biostructures GmbH generated the co-crystal structure of the alpha-3 domain of MICA and CLN-619. Figure 2 was created with Biorender.com.
References
Footnotes
Contributors KAW and CH wrote the manuscript, with support from NKM, KM, KR, PAB, and JSM. Experiments were planned by KAW, CH, NKM, KR, NG and SY. JSM was the guarantor for the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests KAW, CH, NKM, KR, KM, PAB, SY and JSM have ownership in Cullinan Therapeutics, which is seeking to commercialize CLN-619.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.