Article Text

Abstract
Background A positive family history, germline mutations in DNA mismatch repair genes, tumours with high microsatellite instability, and loss of mismatch repair protein expression are the hallmarks of hereditary non-polyposis colorectal cancer (Lynch syndrome). However, in ∼10–15% of cases of suspected Lynch syndrome, no disease-causing mechanism can be detected.
Methods Oligo array analysis was performed to search for genomic imbalances in patients with suspected mutation-negative Lynch syndrome with MLH1 deficiency in their colorectal tumours.
Results and conclusion A deletion in the LRRFIP2 (leucine-rich repeat flightless-interacting protein 2) gene flanking the MLH1 gene was detected, which turned out to be a paracentric inversion on chromosome 3p22.2 creating two new stable fusion transcripts between MLH1 and LRRFIP2. A single-nucleotide polymorphism in MLH1 exon 8 was expressed from both alleles, initially pointing to appropriate MLH1 function at least in peripheral cells. In a second case, an inherited duplication of the MLH1 gene region resulted in constitutional MLH1 promoter methylation. Constitutional MLH1 promoter methylation may therefore in rare cases be a heritable disease mechanism and should not be overlooked in seemingly sporadic patients.
- Lynch syndrome
- gene rearrangement
- hereditary colorectal cancer
- sequence inversion
- cancer: colon
- microarray
- epigenetics
- molecular genetics
Statistics from Altmetric.com
- Lynch syndrome
- gene rearrangement
- hereditary colorectal cancer
- sequence inversion
- cancer: colon
- microarray
- epigenetics
- molecular genetics
Introduction
Hereditary non-polyposis colorectal cancer (HNPCC) is the most common autosomal dominant predisposition to early-onset colorectal cancer (CRC) and increased risk of associated tumours in endometrium, stomach, small intestine, hepatobiliary system, ureter, renal pelvis, ovary, brain and skin1 (MIM 114500). The molecular diagnosis of HNPCC, also known as Lynch syndrome, is the detection of a pathogenic germline mutation in one of the DNA mismatch repair (MMR) genes MLH1 and MSH2, more rarely in MSH6, and to a lesser extent in PMS2. Suspicion of HNPCC is raised according to the Amsterdam criteria2 3 or the less stringent Bethesda guidelines.4 Depending on the HNPCC selection criteria, up to 70–80% of cases can be solved and categorised as Lynch syndrome due to pathogenic mutations, 15% carry unclassified sequence variants such as missense mutations with unknown pathogenicity, but, in 10–15% of cases of suspected Lynch syndrome, no disease-causing mechanism can be detected. Therefore further pathomechanisms are assumed to exist for these genes (for a review, see Cooper et al5).
Deletions and duplications affecting exonic regions in one of the MMR genes, which—except for partial exon deletions/duplications—are easy to detect by multiplex ligation-dependent probe amplification (MLPA), have been commonly found in MSH2 and MLH16–15 and rarely in MSH6.16 Genomic rearrangements, such as deletions and insertions within intronic regions or flanking the gene in question, are difficult to both detect—for example, by Southern blot analysis9 11 13 17 or custom-made zoom-in arrays18 19—and interpret.
So far, only one inversion involving MSH2 exon 1–7 is known to be a disease-causing mechanism in Lynch syndrome.20 21 For MSH2, a transcriptional silencing mechanism is induced by deletion of the last exon of the EPCAM/TACSTD1 gene located upstream of MSH2, leaving the MSH2 gene and promoter intact.17 22 23 If at least EPCAM exon 9, including the termination signal and polyadenylation site and the 3′-UTR, is deleted, a fusion transcript between EPCAM and MSH2 is generated. Depending on EPCAM expression, this read-through transcript into MSH2 silences normal MSH2 transcription starting from exon 1 and induces MSH2 promoter methylation—for example, in colon mucosa. Such regulatory expression mechanisms may also exist for MLH1, MSH6 and PMS2. Regulatory disturbances, such as antisense transcripts, micro-RNAs, cis- and trans-regulatory elements, upstream open reading frames and copy number variations, are also possible causes of HNPCC predisposition.5
Searching for additional hereditary disease-causing mechanisms in MLH1, we analysed 32 cases of mutation-negative suspected Lynch syndrome with MSI-H and MLH1 negative tumours by comparative genomic hybridisation (CGH) analysis and/or MLPA.
We here report for the first time a paracentric inversion on chromosome 3p22.2 between the DNA MMR gene, MLH1, and the downstream LRRFIP2 gene transcribed in antisense direction, which creates two new stable fusion transcripts, thereby abolishing MLH1 gene and protein function. Furthermore, we report heritable partial MLH1 promoter methylation, which is induced by a large genomic duplication including the complete MLH1 gene, the promoter and neighbouring genes.
Materials and methods
Patients
Twenty-three Amsterdam and nine Bethesda positive patients with CRC negative for MLH1 immunohistochemistry were included if MMR gene sequencing and deletion/duplication screening did not detect any germline mutation. The BRAF mutation p.Val600Glu was excluded in tumours of all nine Bethesda patients. Ten patients from Amsterdam families with MLH1 missense mutation of unclear pathogenicity were also included. We also analysed one patient with a known duplication of the whole MLH1 gene region. Patients were recruited in six centres of the German HNPCC consortium. All patients gave written informed consent for the study, approved by the ethics committees. DNA from peripheral blood cells was extracted using the Flexigene Kit (Qiagen, Hilden, Germany), and DNA extraction of tumour tissue and normal tissue from paraffin-embedded material was achieved by microdissection.
Genomic situation
The genomic situation was analysed by oligo array CGH (oligo aCGH; 105K; Agilent, Böblingen, Germany) and MLPA kits (P008, P003, P248, ME011). MLPA analyses were performed following the manufacturer's instructions. The methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA kit ME011, MRC-Holland, Amsterdam, The Netherlands) method quantifies the copy number and methylation in five CpG dinucleotides of the MLH1 promoter in DNA from blood or tumour. In MS-MLPA, ligation of MLPA probes is combined with digestion of genomic DNA with the methylation-sensitive endonuclease HhaI and calculated against the undigested MLPA assay.
Abnormalities such as expected genomic breakpoints were investigated by PCR with Expand Long-Range (Roche, Penzberg, Germany) in a touch-down PCR programme over 64–50°C allowing amplification of fragments of 2–8 kb, which were then sequenced.
cDNA analysis
Total RNA was extracted from peripheral blood for all patients of the study cohort using the PAX Gene Blood RNA and Preparation Kit (PreAnalytiX, Hombrechtikon, Switzerland). RNA isolation after incubation with and without emitine in cell culture to block/not block nonsense-mediated mRNA decay (NMD) was carried out only for the index patient of the family harbouring the inversion. cDNA was generated with the First-strand cDNA-Synthesis Kit (Amersham Biosciences, Freiburg, Germany). Heterozygosity analysis of single-nucleotide polymorphism (SNP) rs1799977 (c.655A→G; p.Ile219Val) in exon 8 of MLH1 was performed with different primers in a standard procedure using Ampli-Taq Gold (ABI, Munich, Germany) or LongAmp DNA polymerase (NEB, Ipswich, UK) followed by sequencing.
For amplification of fusion transcripts, primers in LRRFIP2 exon 1 or 3 forward and MLH1 exon 19 reverse and MLH1 exon 13 forward and LRRFIP2 exon 29 reverse were used. Primer sequences used to analyse potential fusion transcripts with other flanking genes (TRANK1, EPM2AIPI, GOLGA4) are available on request.
Results
Case 1
Genomic situation at a first glance
Of the 31 patients with unexplained loss of MLH1 expression in the CRC, oligo array analyses were performed in 17 patients detecting a deletion in LRRFIP2, a gene located downstream of MLH1 with antisense orientation (figure 1) in one case. The deletion in the region downstream of MLH1 was verified by an MLPA probe in LRRFIP2 exon 26, whereas MLH1 exon 1–19 and its 3′-UTR 128 nucleotides after the termination codon were unaffected.

Schematic genomic organisation of MLH1 and LRRFIP2. The deletion detected by multiplex ligation-dependent probe amplification (MLPA) probes and oligo array probes are depicted as arrows (in black for normal; in grey for 50% reduced signal intensity). Maximal and minimal deletion estimates are shown in grey boxes. Transcription directions of the genes are indicated by arrows within the line constituting the genomic region of the genes; exons are depicted as vertical bars.
A deletion starting downstream of the MLH1 termination codon affecting LRRFIP2 could itself not explain the pathogenicity of the MLH1 gene.
cDNA analysis
To further clarify this finding, mRNA expression analyses were performed for the patient using a heterozygous coding SNP c.655A→G; p.Ile219Val in MLH1 exon 8. In cDNA isolated from the PAX gene, the SNP in MLH1 showed biallelic expression by amplifying exons 3–9, 6–9, 3–11, 7/8–14, but monoallelic expression of the c.655G allele in PCR fragments from exon 1–19, 7/8–16, and 7/8–18. The cDNA with emitine incubation before RNA isolation to block NMD showed the same results with monoallelic expression of the c.655G allele of MLH1 in Long-Range PCR from exon 1–19. Biallelic expression seemed to be restricted to mRNA fragments harbouring MLH1 exon 1–14, and seemed to be monoallelic in full-length MLH1 transcripts.
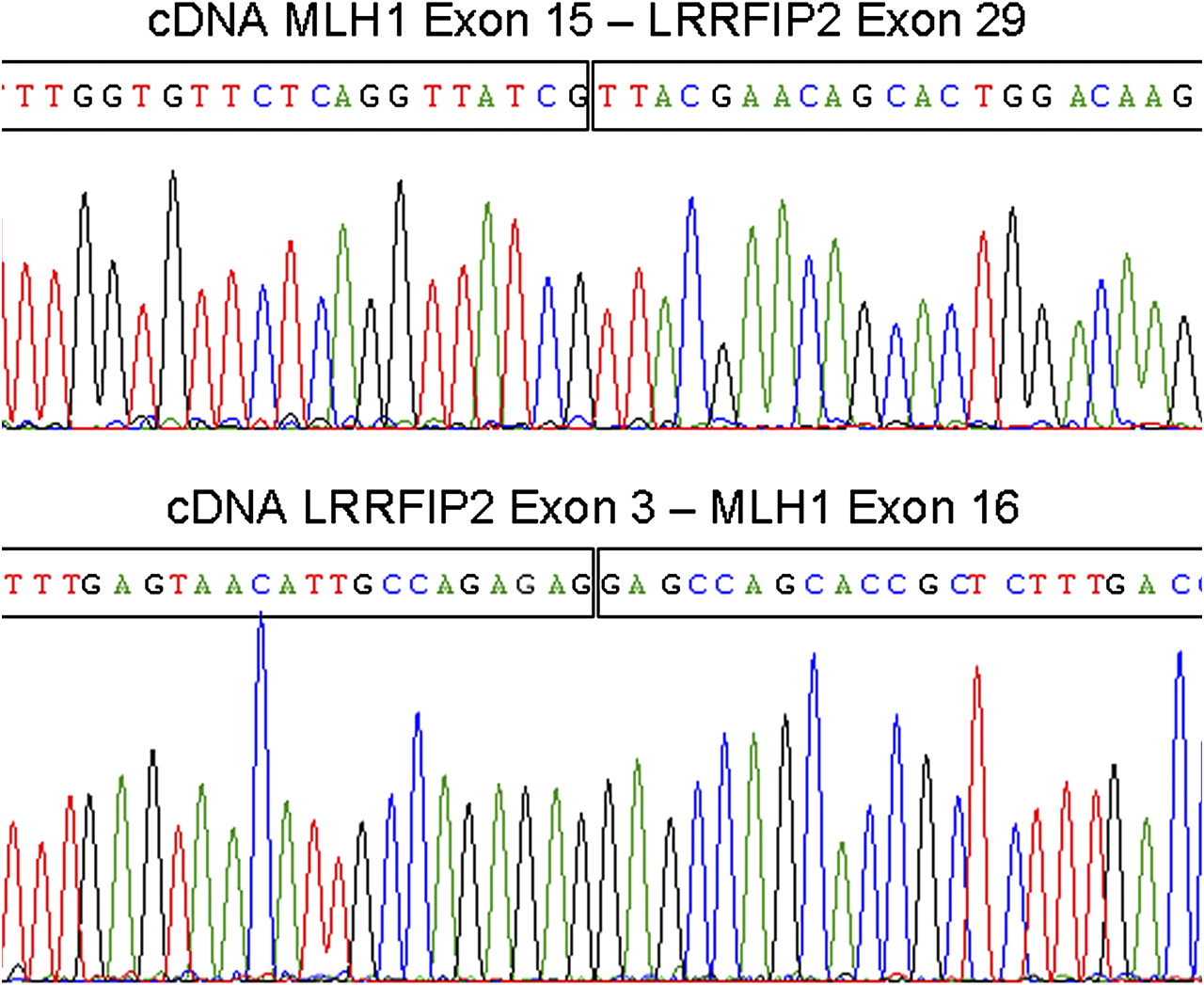
Combining the genomic deletion in LRRFIP2 and the monoallelic MLH1 expression in cDNA analysis beyond exon 14, we suspected a paracentric inversion with one breakpoint in the genomic region of MLH1 and the other breakpoint downstream of MLH1, possibly in the deleted region of LRRFIP2. Further cDNA analyses identified two fusion transcripts, one of MLH1 exon 1–15 fused to LRRFIP2 exon 29 in-frame, and the other one of LRRFIP2 exon 1–3 fused with MLH1 exon 16–19, also in-frame (figure 2).

Sequences of fusion transcripts between MLH1 and LRRFIP2 in cDNA analyses. MLH1 exon 1–15 and LRRFIP2 exon 29 (top) and LRRFIP2 exon 1–3 and MLH1 exon 16–19 (bottom). In-frame fusion points are depicted by vertical lines within the sequences.
Genomic situation at a second glance
The inversion breakpoints in MLH1 intron 15 and LRRFIP2 intron 3 with deletion of 93.751 bp in LRRFIP2 comprising exons 4–28 were characterised in genomic DNA by Long-Range PCR and sequencing (figure 3A). The breakpoint in MLH1 is located after exon 15 in intron 15 c.1731+2148 and is fused to LRRFIP2 c.2056-221 within intron 28 of LRRFIP2, so that MLH1 exon 1 to exon 15 is now in line with LRRFIP2 exon 29 (last exon). The remaining 3′ sequence of MLH1 from intron 15 c.1731+2156 towards the 3′-ends within the inverted fragment fused to LRRFIP2 exon 1 to exon 3 c.90+1096 with sequence loss of seven nucleotides, c.1731+2149_2155TACTTGA, in intron 15 and an insertion of five nucleotides, ATGGT, between the breakpoints, so that the genomic sequence of LRRFIP2 exon 1 to exon 3 is now in line with MLH1 exon 16 to exon 19 (figure 3B). The breakpoint within MLH1 intron 15 is localised within an AluSz sequence motif; the sequence of the breakpoint in LRRFIP2 cannot be predicted because of the large deletion of 93 751 bp of the genomic sequence of LRRFIP2 from intron 3 c.90+1097 to intron 28 c.2056-222. A micro-homology of the sequence of CAGGT was shared between MLH1 intron 15 and LRRFIP2 intron 28 at the fusion point.

(A) Genomic structure of the paracentric inversion. Top line: original genomic structure of MLH1 and LRRFIP2; exons are depicted in arrows according to their transcriptional orientation, UTRs in boxes, MLH1 in dark grey and LRRFIP2 in unfilled (white) boxes and arrows. Second line: the deleted region is represented by a box, filled in light grey. Third line: genomic structure after inversion with breakpoints in MLH1 intron 15 and LRRFIP2 intron 3 and deletion of LRRFIP2 exons 4–28. Bottom line: the stable fusion transcripts of MLH1 exon 1–15 and LRRFIP2 exon 29 and in antisense direction LRRFIP2 exon 1–3 and MLH1 exon 16–19 both in-frame. (B) Sequence analysis of the genomic fusion points of the inversion in MLH1 intron 15 and LRRFIP2 intron 28 (top) and LRRFIP2 intron 3 and MLH1 intron 15 (bottom). In MLH1 intron 15, seven nucleotides were lost (red), and five nucleotides (highlighted) were inserted within the fusion points of LRRFIP2 intron 3 and MLH1 intron 15; in LRRFIP2, intron 3 to intron 28 were deleted.
Clinical data and further screening
The inversion was detected in the index CRC patient of a large family (figure 4) fulfilling the Amsterdam criteria and segregated with CRC and/or endometrial cancer, with ages of first tumour diagnosis ranging from 26 to 52 years in a further three family members. Strikingly, one female without the inversion was diagnosed as having ovarian cancer at the age of 47 years, which is not one of the major tumours in the HNPCC spectrum, especially not in MLH1 carriers, and a sigmoid colon cancer at the age of 56 years. As the inversion segregates with the other affected family members, the tumours in this patient without MLH1 inversion were judged to be coincidental sporadic tumours.

Family pedigree of case 1 with (INV+) and without (INV−) the MLH1 inversion carriers and their ages at tumour diagnosis in years. asc, colon ascendens; biallic, biallelic; ca, cancer; CRC, colorectal cancer; expr, expression; IHC, immunohistochemical staining; monoallic, monoallelic; P8, MLH1 exon 8 single-nucleotide polymorphism.
We screened 12 cDNA samples from patients with suspected Lynch syndrome with unclear MLH1 deficiency for fusion transcripts of MLH1 and LRRFIP2 in one setting with primers for MLH1 exon 1 forward and/or MLH1 exon 11 forward in combination with primers for LRRFIP2 exon 29 reverse. In a second setting, primers for LRRFIP2 exon 2 forward and MLH1 exon 19 reverse were used. These Long-Range PCR settings allow amplification of different fusion transcripts between the two genes up to 1.5 kb in size. Aside from the fragment amplified in the patient with the known inversion, no other fusion transcripts were detected in other patients or controls. Of note, as NMD destabilises transcripts harbouring a premature stop codon at least before 55 nucleotides before the last exon–junction complex, novel stable transcripts are expected only in the case of in-frame fusion transcripts. Patients with out-of-frame fusion transcripts between these two genes would probably have been missed by this approach, as we had only PAX RNA without a block of NMD for these patients.
Case 2
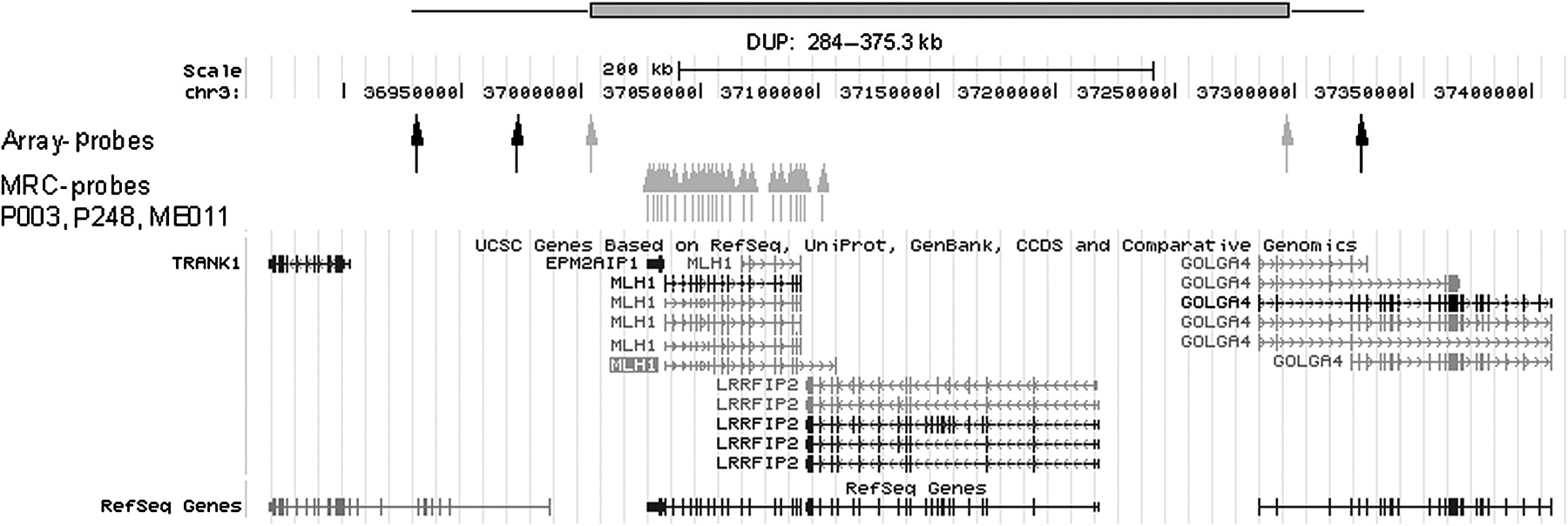
A large duplication of the complete MLH1 gene from exon 1 to exon 19 including the promoter region was found in another patient with CRC by MLPA, leaving the gene itself intact. CGH analysis showed that the duplication started 5′ of the TRANK1 (LBA1) gene completely encompassing EPM2AIP1, MLH1 and LRRFIP2, and the 5′ part of GOLGA4 (figure 5). The duplication (minimal size 280 kb, maximal size 375 kb) is located intrachromosomally (3p22.2), as shown by fluorescence in situ hybridisation with three probes covering the genomic region of MLH1 (as published previously24). The MLH1 duplication was identified in the index patient presenting with colon transversum cancer at 39 years of age. His mother was sent for colonoscopy after molecular testing revealed a carrier status, where she was diagnosed as having CRC at the age of 65. His sister was unaffected at the age of 44 years, and her twin brother did not carry the duplication (figure 6).

Schematic genomic organisation of the duplication region including the EPM2AIP1, MLH1 and LRRFIP2 gene and part of GOLGA4 detected by multiplex ligation-dependent probe amplification (MRC) probes and oligo array probes depicted as arrows (in black for normal; in grey for duplicated signal intensity). The minimal duplication is shown as a red bar. Transcription directions of the genes are indicated by arrows within the line constituting the genomic region of the genes, and exons are depicted as vertical bars.

Family pedigree of case 2 depicting the family members with duplication and constitutional methylation for MLH1 (Meth.+) in peripheral blood cells and their disease status. CRC, colorectal cancer.
All duplication carriers revealed an MLH1 promoter hypermethylation of five MLH1 probes of the MS-MLPA kit from 8% to 18% or 14% to 25% in DNA from blood (figure 7), hair follicle, colonic and buccal mucosa. As no coding SNP in MLH1 or EPM2AIP1 was identified, quantification of MLH1 expression could not be investigated in cDNA. Furthermore, no fusion transcripts between TRANK1, EPM2AIP1, MLH1, LRRFIP2 and GOLGA4 were detectable by PCR approaches using primers in the above mentioned neighbouring genes and MLH1. However, no cDNA with blocked NMD is available so far.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) comparison of methylation-insensitive MS-MLPA ligations (upper lines) analysing copy number, with all MLH1 promoter probes showing the duplication (black arrows) in the patient and the methylation-sensitive MS-MLPA digestion in the patient and control (bottom lines): in addition to undigested control fragments (one methylation-insensitive MLH1 promoter probe indicated by blue arrow shows the duplication), five methylation-sensitive probes of the MLH1 promoter (red arrows) reveal a level of a ∼10% methylation for the patient DNA.
Discussion
We report a disease-causing rearrangement mechanism, which at first glance appeared to be a deletion of the LRRFIP2 gene downstream of MLH1, but was revealed to be a paracentric inversion between the two genes with one breakpoint in MLH1 intron 15 and the other breakpoint in LRRFIP2 intron 3, creating two new in-frame fusion transcripts between these two genes (figure 3A).
The inversion was found in an Amsterdam criteria positive family, in four affected family members whose CRC was MSI-H and negative for MLH1/PMS2 staining. After negative mutation analysis of MLH1 and PMS2, expression analysis by testing small cDNA fragments including a SNP in MLH1 exon 8 also produced normal results, indicating that we had not missed a truncating mutation or a severe regulatory defect. aCGH revealed a deletion in the flanking gene, LRRFIP2, pointing to a possible genomic rearrangement. Only very detailed cDNA analysis deciphered monoallelic expression of the 3′ part of the MLH1 transcript and fusion transcripts with the flanking LRRFIP2 as the disease-causing mechanism. The inversion is therefore assumed to be predisposing for Lynch syndrome.
It is now well established that repetitive DNA sequences, such as Alu repeats, can act as facilitators of chromosomal rearrangements,25 as seems to be the case for the breakpoint in MLH1 intron 15 located in an AluSz. The presence of a microhomology sequence of 5 nucleotides at the breakpoint junction MLH1 intron 15 c.1731+2144_2148 and LRRFIP2 intron 28 c.2056-222_−226 may indicate a homology-based joining mechanism. As LRRFIP2 has a large deletion of 93.7 kb including several Alu repeats, the original breakpoint potentially involving an Alu repeat cannot be characterised.
We assume that the fusion of MLH1 exon 1–15 with LRRFIP2 exon 29, and LRRFIP2 exon 1–3 with MLH1 exon 16–19 would abolish the protein function of both proteins. The leucine-rich repeat flightless-interacting protein 2 encoded by LRRFIP2 is ubiquitously expressed in all tissues, with higher expression in heart and skeletal muscle. It may function as an activator of the Wnt signalling pathway, in association with DVL3, upstream of CTNNB1/β- catenin.26–29 So far, no mutations in LRRFIP2 have been described in association with human disease. Besides CRC predisposition (Lynch syndrome) caused by the defective MLH1 gene, no other disease phenotype was obvious in the affected family described here that would have been attributable to LRRFIP2 haploinsufficiency. No further in-frame fusion transcripts between the two genes were detected by analysing cDNAs of 12 patients with suspected Lynch syndrome with unclear MLH1 deficiency. Out-of-frame fusion transcripts would have been missed, as no NMD-blocked cDNA was available for these patients. So far, only one disease-causing rearrangement resulting in a fusion transcript of MLH1 exons 1–11 with ITGA9 exons 17–28 in-frame has been described, which was generated by an interstitial deletion of about 400 kb on chromosome 3p21.3 in a Lynch syndrome family.30
Partial duplications of MLH1 or MSH2 have been described,6 13 14 19 31 but they never affected the complete genomic region. We describe here a large duplication involving the complete MLH1 gene, the promoter region and flanking genes, leaving the gene itself intact. It was initially found in a seemingly sporadic patient, but was also present in his affected mother (diagnosed after surveillance colonoscopy was recommended) and unaffected sister. All duplication carriers had a low constitutional MLH1 promoter hypermethylation of 10–15% detectable in different tissues. Hypermethylation in these patients was assumed to be due to the duplication of MLH1 and its flanking region, induced by a so far unknown mechanism.
Isolated deletion of MLH1 exon 1 and 2 resulted in one case in constitutional promoter methylation and transcriptional silencing of the gene.32 Promoter hypermethylation of MLH1 therefore seems to be a rare event in cases of heritable genomic rearrangements of the MLH1 gene.
What lessons need to be learnt? Testing on biallelic SNP expression in peripheral cells has been proposed as a large-scale screening method and is a widely used tool to further clarify the situation in patients without detectable germline mutation in the supposedly affected gene.33 However, generation of fusion transcripts can mimic normal biallelic expression and hide genomic rearrangements or imbalances. Thorough cDNA analysis therefore needs to cover the complete transcript in one or several cDNA fragments of different size to ensure that pathogenic ‘partial’ expression of the gene is not overlooked.
Analysis of the MLH1 promoter methylation in seemingly sporadic patients (case 2) is usually, in combination with BRAF mutation analysis, restricted to DNA samples from the tumour to sort out sporadic cases. To a far lesser extent, methylation of the MLH1 promoter was found to be positive in the tumour and in peripheral cells, again in sporadic patients.32 34–38 As long as the mechanistic background remains unclear, aberrant MLH1 promoter methylation can be assumed to be the disease-causing mechanism (in the sense of a de novo constitutional epimutation) and considered not to be inherited by the next generation because of reprogramming in the germline. However, in the literature, one case of maternal transmission of the constitutional MLH1 epimutation to one son with 42% methylation in blood and 0% methylation in sperm has been described, while the other two allele-sharing sons were methylation negative.37 Promoter methylation can also be regarded as an indicator of MLH1 silencing by other causes in cis (elements on DNA are elements in the vicinity of the gene in question, trans elements can be on another chromosome), which may be heritable, especially if found at low methylation levels. In the present case, a low degree of methylation was present in peripheral blood cells and was probably due to a large genomic rearrangement. A positive family history was revealed after genetic testing and surveillance colonoscopy. Another example is mosaic MSH2 promoter methylation, as a result of expression-dependent read-through of EPCAM transcription into MSH2, thereby inducing transcriptional silencing of MSH2 in EPCAM-deletion patients.22 23 Furthermore, a partial duplication of PTPRJ results in silencing due to a read-through of duplicated exon 1–11 into exon 2–3 of the intact gene causing promoter methylation.39
We do not yet routinely look for large genomic rearrangements in these mostly sporadic patients with MLH1 methylation in the tumour without BRAF mutation to further clarify disease mechanisms and define heritability. Large genomic rearrangements other than exonic deletions or duplications are difficult to detect, especially inversions without large deletions. In a previous study, we ruled out translocations and large inversions by cytogenetic analysis (fluorescence in situ hybridisation) in 13 patients with MLH1-negative tumours.24
The finding of abnormal results—that is, duplications or deletions—in only a few probes of an oligo aCGH must be verified with other methods and interpreted with caution. Deletions and insertions flanking the gene in question or located in intronic regions are not detected in routine diagnostics, and it is difficult to judge their pathogenicity, as they may be rare and not necessarily disease-causing, as demonstrated in the following cases. A deletion far upstream of MLH1 was not regarded as disease-causing, as it was reported in a patient also presenting with deletion of MSH2 exon 1–7, as well as the deletion in MSH2 intron 6, which was found in a patient with an additional deletion in MLH1 exon 1.18 Three further aberrations were reported: a deletion in MLH1 intron 13 and a deletion upstream of MSH6 were of unclear pathogenicity, and an insertion in PMS2 intron 7 proved to be pathogenic as it resulted in aberrant splicing.17
Furthermore, regulatory disturbances, such as antisense transcripts, micro-RNAs, cis- and trans-regulatory elements, upstream open reading frames and copy number variations, are also possible causes of HNPCC predisposition.5 In the remaining unsolved cases of suspected HNPCC, we expect further pathomechanisms in the genome to decommission the respective gene, which we hope will soon be detectable by new technologies such as next generation sequencing.
References
Footnotes
Funding This work was supported by the Wilhelm-Sander Foundation and the German Cancer Aid (Deutsche Krebshife).
Competing interests None.
Patient consent Obtained.
Ethics approval Local ethics committees of all university hospitals involved in the study approved the study protocol.
Provenance and peer review Not commissioned; externally peer reviewed.