Article Text

Abstract
Background The microtubule actin crosslinking factor 1 (MACF1) gene encodes microtubule–microfilament cross-linking factor 1 that plays an essential role in the embryonic brain development. MACF1 variants were associated with lissencephaly-9 (LIS9). However, the MACF1-epilepsy relationship was unknown.
Methods Trios-based whole-exome sequencing was performed on a cohort with generalised epilepsy from the China Epilepsy Gene 1.0 project. The spatial–temporal expression, single-cell sequencing and genotype–phenotype correlation were analysed to explore the role of MACF1 in epilepsy and neurodevelopment.
Results Two de novo heterozygous and eight biallelic MACF1 variants were identified in 10 unrelated patients. The variants presented significantly high excess by multiple statistical analyses. All patients were diagnosed with generalised epilepsy, among whom three patients presented with neurodevelopmental delay. MACF1 was expressed throughout the lifespan, with three major peaks in the fetal, early childhood and adulthood stages, consistent with seizure onset ages of the patients. The highest expression in adulthood was in the thalamus nucleus, potentially associated with the pathogenesis of generalised epilepsy. The single-cell sequencing in organoids showed MACF1 is widely expressed in the developing brain, especially in the early stage, suggesting a vital role in neurodevelopment. Genotype–phenotype association analysis revealed that LIS9-associated variants were featured by de novo monoallelic variants clustered within the C-terminal; the autism spectrum disorder-associated variants were mainly de novo monoallelic variants located at the spectrin-repeat rod domains. In contrast, the epilepsy-associated variants were biallelic missense variants, and those in the plakin domain were potentially associated with neurodevelopment delay.
Significance MACF1 is potentially a novel causative gene of generalised epilepsy.
- Genetic Research
- Whole Exome Sequencing
- Gene Expression
- Epilepsy
- Genotype
Data availability statement
Data are available upon reasonable request. The data that support the findings of this study are available in the supplementary material of this article or available from the corresponding author upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Microtubule actin crosslinking factor 1 (MACF1) is vital for neurodevelopment, of which variants have been reported to be associated with autosomal dominant lissencephaly-9 (LIS9) with complex brainstem malformation.
The relationship between MACF1 variants and epilepsy, especially generalised epilepsy, remains unknown.
WHAT THIS STUDY ADDS
This study identified novel MACF1 variants in patients with generalised epilepsy and with/without neurodevelopmental delay (NDD).
MACF1 showed three major expression peaks that were consistent with seizure onset ages of the patients.
Genotype–phenotype analysis revealed that LIS9-associated variants were clustered within the GAS2 domain; autism spectrum disorder-associated variants were located at the spectrin-repeat rod domains; whereas, epilepsy with NDD-associated variants were in the plakin domain.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
MACF1 is potentially a novel causative gene of generalised epilepsy.
The genetic dependent (expression) stage of MACF1, especially high expression in the adulthood stage, suggests attention on the long-term treatment and outcome of the patients.
Introduction
Epilepsies are common neurological disorders with high clinical heterogeneity, which is generally categorised into focal and generalised epilepsy. Focal epilepsy, which arises from abnormal electrical activity in a specific region of the brain, can be caused by a variety of causes, including genetic and acquired factors. Generalised epilepsy, which presents a lifetime prevalence of 4.33/1000,1 is characterised by the simultaneous involvement of both cerebral hemispheres and is largely believed to be genetically determined. Currently, 2946 genes were reported to be potentially associated with epilepsy,2 among which only about 20 genes were found to be causative/susceptibility for generalised epilepsy, such as CLCN2, GABRA1, GABRB3 and SLC2A1;2 3 and some genes with questionable epileptogenic effects such as CACNA1H and EFHC1.4 5 Our recent studies have identified USP256 and CSMD17 as novel causative genes of generalised epilepsy. However, the genetic causes in most patients with generalised epilepsy remain to be elucidated.
MACF1 (OMIM *608271, also known as ACF7) encodes microtubule actin crosslinking factor 1 (MACF1), which is ubiquitously expressed across the whole lifespan, especially in the embryonic nervous system.8 Variants in MACF1 have been reported to be associated with autosomal dominant lissencephaly-9 with complex brainstem malformation (LIS9, OMIM #618325),9 characterised by global developmental delay, severe intellectual disability and congenital brain malformation. Generalised seizures were occasionally observed in patients with LIS9.9 10 However, the association between MACF1 variants and epilepsy, especially generalised epilepsy, remains unknown.
In this study, trios-based whole-exome sequencing (WES) was performed in a cohort of patients with generalised epilepsies, a subcohort recruited from the China Epilepsy Gene 1.0 project. De novo heterozygous missense and compound heterozygous missense variants in MACF1 were identified in 10 unrelated patients with generalised epilepsies, among whom 3 patients presented with neurodevelopmental delay (NDD). The MACF1 gene presented significantly higher excesses of variants in the epilepsy cohort than that in the controls. The spatiotemporal expression pattern and the single-cell expression of MACF1 in cerebral organoids were analysed to explore the underlying mechanisms of generalised epilepsy and NDD. The genotype–phenotype correlation was investigated to explore the gene-disease association of MACF1. This study suggested that MACF1 is potentially a novel causative gene of generalised epilepsy.
Materials and methods
Subjects
The patients were recruited from the ongoing China Epilepsy Gene 1.0 Project, which was categorised into 16 subcohorts according to the clinical phenotypes. A total of 340 patients, from the subcohort of generalised epilepsy, were recruited in this study from 2016 to 2023. Epileptic seizures and epilepsy syndromes were diagnosed according to the criteria of the Commission on Classification and Terminology of the International League Against Epilepsy (1989, 2001, 2010, 2017 and 2022). Generalised seizures were diagnosed based on a range of seizure types including absence, spasms, myoclonus, clonic, atonic, tonic and tonic-clonic seizures, supported by generalised discharges on EEGs.
The clinical information of patients was collected, including the age at seizure onset, the type and frequency of seizures, the family history, the response to antiepileptic medication and the results of the general and neurological examinations. The development of motor, language and intelligence was assessed. MRI scans were performed to identify brain abnormalities. Video-EEG monitoring was conducted and reviewed by two qualified electroencephalographers. Patients with acquired causes, such as hypoxic–ischaemic encephalopathy, central nervous system infections and cerebral trauma, were excluded. All the enrolled patients were followed up for at least half a year.
Whole-exome sequencing and genetic analysis
Blood samples were obtained from the probands and their biological parents (trios). Genomic DNAs were extracted from the blood samples using the Qiagen FlexiGene DNA kit (Qiagen, Hilden, Germany). Trio-based WES was performed on the NextSeq 500 sequencing instruments (Illumina, San Diego, California, USA). The exome capture, sequence alignment and variant calling were performed according to the standard procedures as previously described.3 11 12
To identify potential pathogenic variants in each case, a tailored strategy was applied.7 11 Initially, rare variants with a minor allele frequency (MAF) <0.005 in the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/) were prioritised. A filter was applied to retain variants with potential pathogenicity, such as non-sense, frameshift, canonical splice site, initiation codon and missense variants. The variants were further filtered based on their explainable inheritance patterns within the trio, focusing on de novo, homozygous, compound heterozygous and hemizygous variants. Subsequently, stratified MAF criteria were applied: de novo, hemizygous and homozygous variants had to be absent in control populations in gnomAD, while for compound heterozygous variants, the product of the allele frequencies needed to be <1×10−6, which is substantially lower than the expected probability of such an occurrence in the gnomAD population (1/141456=7 × 10−6). Ultimately, the candidate genes were refined using stringent criteria derived from the Genetic Dependence & Pathogenicity Database (www.gdap.org.cn), which comprehensively encompassed four pivotal facets of the gene profiles:
Tissue-specific expression: a prerequisite for considering a gene as epilepsy-causing is its expression within the brain (inclusion criterion). Alternative possibilities, such as ectopic expression or indirect toxic effects from abnormal metabolic byproducts, were also considered.
Exclusion of conflicting gene-disease associations: genes that have been definitively linked to diseases incompatible with epilepsy phenotypes were systematically excluded (exclusion criterion).
Assessment of gene intolerance to variants: the probability of a gene being intolerant to heterozygous or homozygous loss-of-function (LoF) variants (pLI/pRec) was evaluated. Special attention was given to genes with a pLI score ≥0.9, harbouring dominant variants, and those with both pLI and pRec scores≥0.9, along with a pNull score ≤0.1 in the context of recessive variants.
Phenotypic consequences of genetic manipulation: the impact of genetic knockout or knockdown on brain-related phenotypes was assessed to further validate the pathogenicity of the genes.
Following the application of the filtering criteria, genes with recurrent variants were prioritised for further analysis to elucidate their specific gene-disease associations. Among these, the MACF1 gene appeared as a prominent candidate because of the repeated occurrence of de novo and biallelic variants within the cohort. All the candidate MACF1 variants were validated by Sanger sequencing and annotated based on the reference transcript NM 012090.5.
Damaging effect of MACF1 variants
To evaluate the damaging effect of candidate variants on protein structure, protein modelling was performed by using the Iterative Threading ASSEmbly Refinement software (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). PyMOL V.1.7 was used for three-dimensional protein structure visualisation and analysis.
To evaluate the effect of missense variants on protein stability, the free energy change value (DDG, Delta-Delta-G, kcal/mol) was predicted by the I-Mutant server.13 A value of >0.5 kcal/mol implies an increase in protein stability, while a value of ≤0.5 kcal/mol is considered to decrease the protein stability, and others imply neutral stability.
Excess of MACF1 variants
The frequencies of MACF1 variants were also compared with those of ‘benign’ variants obtained from the gnomAD database. To assess the effect of allele frequencies on disease, the aggregate frequency of variants in the cohort with epilepsy was compared with the general populations of gnomAD.14 To analyse the significance of the compound heterozygous variants, we established a control cohort that included 1942 asymptomatic parents from trios, in whom the compound heterozygous variants were identified by detecting one of the paired variants in the child, given that one of the paired variants in a parent would transmit to the child. The significance of the compound heterozygous variants was also compared with the data from the gnomAD database.
Spatial and temporal expression analysis
To analyse the effect of the spatial-temporal expression of MACF1 on disease, the MACF1 expression pattern at different tissues and different developmental stages was analysed. The MACF1 expression profiles across organs and developmental stages were calculated with data from the Evo-devo mammalian organs database. Expression in adult non-disease tissues was analysed with data from the GTEx database. Bulk RNA-seq data of developmental expression in the brain were analysed using the BrainSpan database.
Single-cell expression of MACF1
The expression of MACF1 across developmental stage was studied via data from developing cerebral organoids and the adult non-disease human brain. Single-cell MACF1 expression data from 1/5-month undirected cerebral organoids were retrieved from the Human Organoid Single-Cell Browser.15 Single-cell data of the adult human brain were obtained from the ScApeX database.16
Statistical analysis
R statistical software (V.4.0.3) was used for statistical analysis. Fisher’s exact test was applied to assess the allele frequencies of MACF1 variants between the case cohort and the benign variants. Two-sided Fisher exact test, as recommended by ClinGen, was employed to compare the aggregate frequency of MACF1 variants between the epilepsy group and the control group. Fisher’s exact test was employed to analyse the difference in the proportion of monoallelic missense variants among varied phenotypes. The Mann-Whitney test was applied to analyse the difference of allele frequencies among varied phenotypes. A p value of<0.05 was statistically significant.
Results
Identification of MACF1 variants
MACF1 variants were identified in 10 unrelated cases with generalised epilepsies (figure 1A and table 1), including 2 de novo heterozygous missense variants (c.9905A>G/p.Asn3302Ser and c.14740C>T/p.Arg4914Cys), and 8 pairs of compound heterozygous missense variants (c.1556T>C/p.Leu519Ser & c.11118T>A/p.Asp3706Glu, c.3014G>A/p.Arg1005His & c.7160A>G/p.His2387Arg, c.3698G>A/p.Arg1233His & c.2908G>T/p.Val970Leu, c.5503G>A/p.Glu1835Lys & c.8498A>T/p.Gln2833Leu, c.6592A>T/p.Asn2198Tyr & c.6245A>G/p.Gln2082Arg, c.9617T>C/p.Leu3206Ser & c.9292G>A/p.Glu3098Lys, c.14785C>A/p.Leu4929Met & c.6245A>G/p.Gln2082Arg and c.16180G>A/p.Ala5394Thr & c.12404C>T/p.Ala4135Val). The biallelic variants were inherited from asymptomatic parents, consistent with an autosomal recessive pattern.

Genetic data of the cases with MACF1 variants. (A) Pedigrees and DNA sequencing chromatograms of the 10 cases with MACF1 variants. (B) Hydrogen bond and free energy (DDG, Kcal/mol) changes of the variants identified in this study. The proteins with hydrogen bond changes or DDG ≤0.5kcal/mol were highlighted in red. DD, developmental delay; DDG, Delta-Delta-G; GE, generalised epilepsy; MACF1, microtubule actin crosslinking factor 1.
Clinical features of the patients with MACF1 variants
No similar MACF1 variants were identified in the subcohort of focal epilepsy in the China Epilepsy Gene 1.0 Project yet.
None of the 10 affected patients had other pathogenic or likely pathogenic variants in the genes known to be associated with epileptic phenotypes.2 17
The damaging effects of the variants were evaluated by hydrogen bonding and protein stability changes (figure 1B). 12 missense variants showed alteration of hydrogen bonds with surrounding residues, 4 variants showed significantly decreased protein stability and 2 variants did not change hydrogen bonding or protein stability. All the missense variants identified in this study were predicted to be damaging by at least two in silico tools (online supplemental table S1).
Supplemental material
Excess of variants
Two missense variants p.Arg4914Cys and p.Leu3206Ser were absent in the gnomAD-all population, and the remaining 16 variants presented extremely low frequencies (MAF<4.8 × 10−4; Online supplemental table S1). Among the compound heterozygous variants, the product of the frequencies of the two alleles in gnomAD is <1×10−6, which is less than the probability of observing an individual with such genotype in the gnomAD population (1/141456=7 ×10−6). None of the variants were observed as homozygotes in the gnomAD populations.
The MACF1 variants identified in the cohort with epilepsy presented significantly lower MAFs than the benign variants in the gnomAD-all populations and controls of gnomAD-all populations (p=0.0021 and p=0.0062, respectively; figure 2A,B). The aggregate frequencies of MACF1 variants in the epilepsy cohort were significantly higher than those in the gnomAD-all population and the controls of the gnomAD-all population (p=4.2×10−16 and p=1.6×10−15, respectively; figure 2C). Additionally, the frequency of compound heterozygous variants in this cohort was significantly higher than that in the inner controls and the gnomAD database (8/340 vs 8/1942, p=9.64×10−4; vs 3/141456, p=1.65×10−19; figure 2C).

Excess of MACF1 variant. (A) The MACF1 variants identified in the cohort with epilepsy presented significantly lower MAFs than the benign variants in the gnomAD-all populations. (B) The MACF1 variants identified in the cohort with epilepsy presented significantly lower MAFs than the benign variants in the controls of gnomAD-all populations. (C) The excess of MACF1 variants in the epilepsy cohort compared with the controls. gnomAD, Genome Aggregation Database; MACF1, microtubule actin crosslinking factor 1; MAF, minor allele frequency.
Clinical features of the patients
The clinical features of the 10 patients with MACF1 were summarised in table 1. All the patients were diagnosed with generalised epilepsy. The onset age of seizures ranged from 1 year to 26 years old, with a median onset age of 4.25 years.
Seven patients, including two patients (case 1 and 2) with de novo heterozygous missense variants and five patients (case 6–10) with compound heterozygous missense variants, presented with infrequent generalised tonic-clonic seizures (GTCS). They all became seizure-free and presented with normal development.
Three patients (case 3, 4 and 5) presented multiple and frequent generalised seizures, including GTCS, myoclonic, absence and tonic seizures. Case 3 and 4 had been seizure-free for 1 year and case 5 still suffered from occasional tonic seizures under the treatment of valproate and lamotrigine. All three cases showed NDD with poor speech, and case 4 also presented with autism spectrum disorder (ASD).
EEGs showed generalised discharges in six patients, both generalised and focal discharges in three patients, and normal in one patient with late-onset age (table 1 and figure 3). Brain MRI was normal in all patients.

Representative EEG of the cases with MACF1 variants. (A) Interictal EEG of case 1 showed generalised spike-and-slow waves (SW). (B) Interictal EEG of case 2 showed generalised spikes and SW; and spike in the right temporal focal. (C) Interictal EEG of case 3 showed generalised 3–3.5 Hz SW and polyspike-slow waves. (D) Interictal EEG of case 4 showed generalised 3–4 Hz SW. (E) Interictal EEG of case 5 showed generalised polyspike-slow waves and multifocal SW. (F) Interictal EEG of case 6 showed generalised SW, predominantly in bilateral frontal and occipital regions. (G) Interictal EEG of case 8 showed generalised polyspike-slow waves. (H) Interictal EEG of case 9 showed generalised polyspike and SW, and SW in the bilateral central–parietal–temporal region. MACF1, microtubule actin crosslinking factor 1.
Spatial–temporal expression of MACF1 gene
The spatial–temporal expression profile is an intrinsic nature of a gene, which potentially determines the characteristics of the associated phenotypes. The gene expression profile of MACF1 was investigated to disclose the gene-disease association. MACF1 is widely expressed across various tissues (figure 4A,B). The subspatial expression profile of MACF1 in the brain showed that it was widely expressed in multiple brain subregions, including the cortex, cerebellar hemisphere, hippocampus, amygdala, thalamus and basal ganglia (figure 4A).

The spatial–temporal expression of the MACF1 gene. (A) Expression of MACF1 in the brain and human tissues. MACF1 is widely expressed in multiple brain subregions. (B) Expressional profile of MACF1 across organs in different developmental stages. MACF1 shows the highest expression in the developing brain before newborn. (C–F) The temporal expression pattern of MACF1 in diverse brain regions. MACF1 was expressed throughout the lifespan, with three major peaks in different regions. (G–H) The expression of MACF1 in the cells of single neural rosette-derived cortical organoids at 1 month and 5 months. (I) MACF1 expression in the adult brain via single-nucleus RNA sequencing (snRNA-seq). MACF1 was extensively expressed in various types of neurons, including the inhibitory and excitatory neurons. Ast, astrocyte; CTX, cortex; EN, excitatory neurons; End, endothelial/pericyte; Ex, excitatory neurons; ID, intellectual disability; IN, inhibitory neurons; IP, intermediate neural progenitors; LGE, lateral ganglionic eminence; MACF1, microtubule actin crosslinking factor 1; Mic, microglia; mos, months; NDD, neurodevelopmental delay; NP, neural progenitors; Oli, oligodendrocyte; OPC, oligodendrocyte progenitor cell; pcw, post conception week; RG, radial glial; RPKM, reads per kb per million mapped reads; Unk, unknown; wpc, weeks post conception; y, years; yrs, years.
Further analysis on the temporal expression pattern in the brain showed that MACF1 was expressed throughout the lifespan, with three major peaks (figure 4B,C). The first peak occurred during the fetal stage, which was observed in most developing brain regions (figure 4C–F). The second peak was observed between the infancy and early childhood stages, consistent with the seizure onset ages for most patients in this study (figure 4C). The third peak of MACF1 expression was observed during the late adolescence/adulthood stage, with the highest expression in the mediodorsal nucleus of the thalamus (figure 4F), an important anatomical region strongly associated with generalised epilepsy.
The single-cell expression of MACF1
The single-cell expression in cerebral organoids showed that MACF1 was widely expressed in the developing brain, especially in the early stages, such as the 1-month-old and 5-month-old organoids, suggesting a vital role in neurodevelopment (figure 4G,H). Additionally, MACF1 was extensively expressed in various types of neurons, including the inhibitory and excitatory neurons (figure 4G–I).
Genotype–phenotype correlation
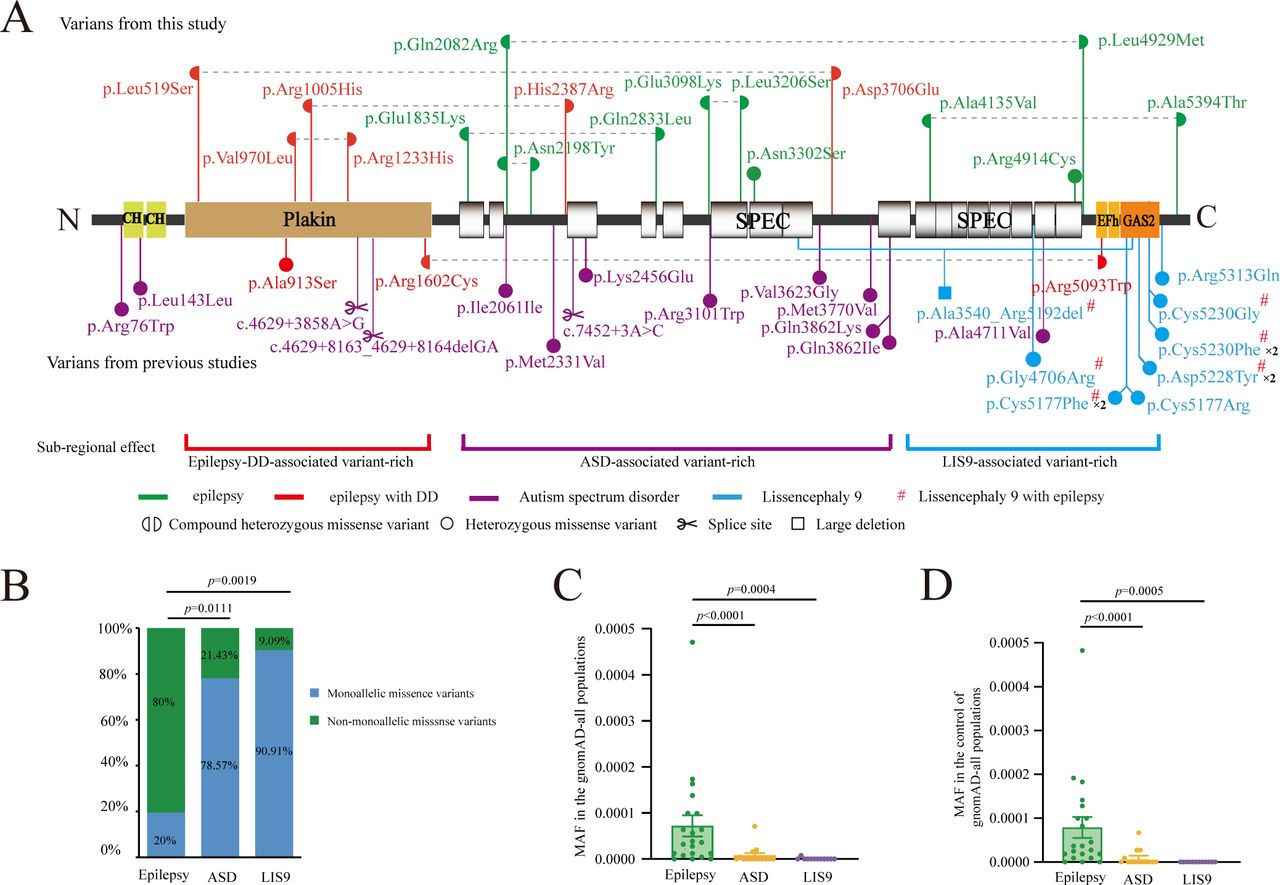
The MACF1 protein consists of an N-terminal domain consisting of actin binding domain and plakin domain, a rod domain composed of spectrin repeats and a C-terminal domain containing EF-motif, GAS2-related protein domain and glycine-serine-arginine domain. The variants identified in this study were illustrated in figure 5A. In the three cases with epilepsy and NDD, the compound heterozygous MACF1 variants had at least one of the paired variants located at the plakin domain. Whereas variants associated with epilepsy without NDD were located within the spectrin-repeat region, suggesting a potential submolecular effect.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic illustration of the variants and MAF-phenotype correlations of MACF1 variants. (A) Schematic diagram of the MACF1 protein and the localisation of the MACF1 variants in this study. (B) The proportion of monoallelic missense variants in cases with epilepsy was significantly lower than those with ASD or LIS9. (C-D) The epilepsy-associated variants showed significantly higher frequency in the gnomAD-all populations (C) and the control of gnomAD-all populations (D) than the ASD and LIS9 associated variants. ASD, autism spectrum disorder; DD, developmental delay; gnomAD, Genome Aggregation Database; LIS9, lissencephaly-9; MACF1, microtubule actin crosslinking factor 1; MAF, minor allele frequency.
To further delineate the genotype–phenotype association, we systematically reviewed previously reported MACF1 variants (online supplemental table S2). MACF1 variants, including one de novo (p.Ala913Ser) and one compound heterozygous variant (p.Arg1602Cys and p.Arg5093Trp), were identified in two patients with features of generalised epilepsy and NDD (figure 5A). Notably, the de novo and one of the paired variants were also located in the plakin domain, consistent with our findings.
Supplemental material
Besides epilepsy, MACF1 variants have previously been identified in other neurodevelopment disorders, including LIS9 with complex brainstem malformation and ASD. Among the patients with LIS9 in the previous studies, two patients present with infantile spasms, two with Lennox-Gastaut syndrome, two with myoclonic seizures and three with mixed focal and GTCS.9 10 The variants associated with LIS9 were clustered within the C-terminal of MACF1, especially in the GAS2 domain; and variants associated with ASD located at the spectrin-repeat region (figure 5A), suggesting a potential subregional implication.
The previously reported ASD-associated and LIS9-associated variants were mainly de novo heterozygous missense. In contrast, in this study, the variants associated with epilepsy were mostly biallelic missense variants. The proportion of monoallelic missense variants in cases with epilepsy was significantly lower than those with neurodevelopmental disorders (vs ASD, p=0.0111; vs LIS9, p=0.0019; figure 5B). The ASD-associated and LIS9-associated variants present lower MAFs in the general populations than the epilepsy-associated variants (p<0.0001 and p<0.0005, respectively; figure 5C,D).
Discussion
In this study, de novo and compound heterozygous MACF1 variants were identified in 10 patients with generalised epilepsy and with/without NDD. These variants presented none or extremely low allele frequencies in the general populations, and the aggregate frequencies were significantly higher than those in the controls. The frequency of compound heterozygous variants in the epilepsy cohort was significantly higher than that in the inner controls and the gnomAD populations. MACF1 is ubiquitously expressed across the whole lifespan, with three major peaks that are consistent with seizure onset ages. Notably, the peak expression in the adult stage is in the mediodorsal nucleus of the thalamus, providing a molecular basis for generalised epilepsy. The high expression of MACF1 in the early developing brain suggested a crucial role in neurodevelopment. The gene-disease association of MACF1 with generalised epilepsy was further supported by the genotype–phenotype correlation analysis. These findings suggest that MACF1 is potentially a novel causative gene of generalised epilepsy.
The MACF1 gene encodes a multidomain cytoskeletal protein of the spectraplakin family,18 19 a scaffolding protein that interacts with both actin microfilaments and microtubules.20 21 MACF1 plays important roles in neuronal migration,22 proliferation,23 neurite arborisation,24 25 and signalling25 26 that are essential for embryonic brain development.27 28 Macf1 knockout mice exhibit embryonic lethality between implantation and somite formation.26 Conditional deletion of Macf1 markedly decreased dendritic branching of cortical and hippocampal pyramidal neurons in the mice developing brain,24 suggesting that MACF1 is vital for neurodevelopment.
Clinically, MACF1 variants have been previously identified in patients with NDD, including LIS99 29 30 and ASD.31 32 The present study identified three of our patients who presented with NDD, including speech delay and ASD. The temporal expression pattern of MACF1 showed that the first peak occurred during the fetal stage, which was observed in most developing brain regions. The single-cell sequencing also showed that MACF1 was highly expressed in the developing brain, including in the 1-month-old organoids and 5-month-old organoids. These findings supported the pathogenic role of MACF1 in neurodevelopmental disabilities.
Notably, MACF1 was broadly expressed in multiple brain subregions, including the cortex, hippocampus, amygdala, thalamus and basal ganglia. In the infant organoids and adult human brains, MACF1 was also widely expressed in various types of neurons. Especially in the late adolescence/adulthood stage, MACF1 showed a third expression peak with the highest expression in the mediodorsal nucleus of thalamus, a critical region associated with the pathogenesis of generalised epilepsy. These findings provided novel insights into the mechanisms underlying generalised epilepsy and late-onset epilepsy. Our recent studies have shown that the genetic-dependent stage6 33–36 and the development-dependent pattern of neuron specificity37 are associated with the onset age, evolution and outcome of genetic diseases. The third peak of MACF1 expression during late adolescence/adulthood stage suggests attention on the long-term treatment and outcome of the patients.
MACF1 contains several crucial domains, each with distinct functional roles.38 The N-terminal calponin-homology domains facilitate actin binding; the plakin domain plays a role in the protein–protein interactions; the long spectrin-repeat rod domain functions in the flexibility of MACF1; and the C terminus GAS2 domain is responsible for microtubule binding.21 39 The present study showed that LIS9-associated variants were featured by de novo monoallelic variants clustered within the C-terminal; the ASD-associated variants were mainly de novo monoallelic variants located at the spectrin-repeat rod domains. In contrast, the epilepsy-associated variants were biallelic missense variants and the variants associated with both epilepsy and NDD were related to the location in the plakin domain. The submolecular implication of variants further supported the gene-disease association of MACF1.
Homozygous knockout of Macf1 in mice resulted in embryonic lethality with complete penetrance.26 In humans, no homozygous MACF1 null variants were reported in the gnomAD general populations, indicating that complete LoF of MACF1 would be lethality. The probability of being LoF intolerant (pLi) of MACF1 is 1.000, suggesting that MACF1 is highly intolerant to monoallelic null variants. Three monoallelic splice variants of MACF1 had been identified in patients with ASD,31 confirming the pathogenicity of a single null variant. On the other hand, the missense Z score of MACF1 is 3.434, suggesting that MACF1 is highly intolerant to missense variants. The present study showed that the majority of variants in NDD and all variants in epilepsy were missense variants, suggesting the specific role of missense variants in the pathogenicity of the MACF1 gene, especially in epilepsy, as shown in SCN2A40 and our several recent studies.37 41–45 This may be one of the explanations for the presence of biallelic missense variants of MACF1 in patients with epilepsy in this study, on which further studies are required to explore the mechanisms.
In conclusion, this study identified MACF1 as a novel causative gene of generalised epilepsy. The disclosed genotype−phenotype correlation implies significance in the diagnosis and management of the patients with MACF1 variants.
Data availability statement
Data are available upon reasonable request. The data that support the findings of this study are available in the supplementary material of this article or available from the corresponding author upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study adhered to the guidelines of the International Committee of Medical Journal Editors with regard to patient consent for research or participation, and was approved by the ethics committee of the Second Affiliated Hospital of Guangzhou Medical University (2020-hs-49). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The help of patients and clinicians participating in this work is greatly appreciated.
References
Footnotes
X-YLe, M-WZ and HS are joint first authors.
X-YLe, M-WZ and HS contributed equally.
Contributors NH is the guarantor. NH and HM designed the study, administered the project and revised the manuscript. X-YLe, M-WZ and HS contributed to data curation, formal analysis and drafting of the manuscript. WS, X-YLi, C-SW and SL collected and analyzed clinical data and patient samples. B-ML, X-RL, YW, YT and QP examined the patients. JW performed the data analysis and provided technical assistance. W-PL provided critical review and substantially revised the manuscript. All authors have read and approved the final manuscript.
Funding This work was funded by Guangdong Basic and Applied Basic Research Foundation (no.2023A1515010218), Science and Technology Projects in Guangzhou (no.2023A03J1026), Guangzhou Medical Key Discipline Construction Project (2025–2027), the National Natural Science Foundation of China (no.82271505) and the 2025 City-School (Guangzhou) Joint Funding Program (no.2025A03J4405). The funders had no role in the study design, data collection and analysis, and decision to publish or preparation of the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.