Article Text

Abstract
Objective Low-risk pragmatic clinical research such as learning health system research, quality assurance activities and comparative effectiveness studies are approaches to embedding clinical research within routine practice to generate generalisable evidence. Individual written informed consent may present a barrier to the feasibility and inclusiveness of such low-risk clinical research. Within an overarching moral and ethical framework, the form and requirements of consent vary in both clinical practice and research settings according to the context and level of risk. Within some low-risk research settings, waiver of consent may be appropriate.
Analysis We sought to describe contemporary national and international English-language guidelines pertaining to the use and oversight of waiver of consent in clinical research. We identify 14 guidelines including 1 international, 1 regional and 12 national statements, and summarise the principles in each for circumstances in which a waiver of consent is appropriate.
Conclusion While complete international harmonisation of policy may be neither realistic nor necessary, there are numerous unifying concordances suggesting a broad consensus on the approach to waiver of consent research.
- STATISTICS & RESEARCH METHODS
- MEDICAL ETHICS
- Research Design
- Clinical Trial
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
Informed consent for research participation is a cornerstone of modern medical ethics and is an essential feature of randomised research involving novel therapies and technologies. While such studies spearhead progress in patient care, many aspects of the healthcare system are unsuited to individual randomised controlled trial designs or seek to assess the impact of differing treatment strategies or systems on broad patient populations that are often not fully captured by studies using traditional informed consent models. This is most relevant for pragmatic low-risk clinical research which aims to evaluate the effectiveness of interventions in real-world settings, often comparing the relative merits of medical treatments already in common use. Such research is often cited as key to achieving the goal of a ‘learning health system’, in which research is embedded within routine practice and the effectiveness of an intervention, practice or system change is assessed in all participants exposed.1 Comparative effectiveness pragmatic randomised controlled trials (RCTs) are a form of learning health system research, which act within areas of existing variation in clinical practice that are not justified by high-quality evidence or local conditions.2 3 These studies replace arbitrary treatment selection with randomised selection and are essential to improving patient outcomes, especially with respect to institutional or health system practices that are determined outside of, or prior to, individual clinician-patient treatment decisions.
Explanatory randomised trial populations have historically differed substantially from the broader patient population.4–7 These differences stem in part from inclusion and exclusion criteria that are designed both to protect participant safety and to define a population most suited to testing the study hypothesis. However, they also result from the informed consent process itself, which requires participants to understand consent material written in technical and legal style (or have a guardian able to do so), and to be sufficiently engaged in their healthcare so as to volunteer or accept referral for inclusion in research. These factors result in study populations which differ in important ways from the broader patient population, for example, being younger, less comorbid or with a better prognosis. Contrastingly, comparative effectiveness studies and evaluations of health-service interventions are designed for broader real-world settings and will naturally lead to changes in practice that affect all patients. Such studies will be most informative if they include generalisable populations, are sufficiently large and minimise bias. Given the barriers that traditional written informed consent models pose to inclusion of a representative patient population, such studies of practices where patient consent is not sought in clinical practice may warrant a similar waiver of research consent to minimise selection and other biases.8–10 The benefits of aligning consent requirements for practice evaluation with those for the conduct of clinical practice have long been recognised. In 1987, Chalmers et al posited that in the face of uncertainty in clinical research and clinical care, the consent processes required for rigorously conducted real-world studies compared with the lack of such requirements for clinical decision-making were an ethical discordance that impeded practice improvement.11 12 Recommendations for facilitating research and clinical practice integration are through the anchoring of consent requirements in a real-world study with those of the equivalent routine clinical practice setting.13–16
The increasing recognition of the impact of health services on clinical outcomes comes at a time when digital advancements are facilitating research and quality assurance studies that were previously technologically challenging or impossible.2 In this context, there is increasing interest in the utilisation of waiver of consent in low-risk clinical research. While the well-established opt-in consent model has broadly accepted advantages for participant autonomy and safety, researchers, ethics committees and the broader health community may benefit from a distillation of global guidance on situations in which it may be ethically acceptable to consider a waiver of consent. We therefore aimed to provide an overview of national and international statements of guidance on principles that outline when a waiver of consent model could be considered for an individual study.
Methods
This analysis examines current international, regional and national English-language guidelines on ‘waiver of consent’ in clinical research within non-emergency settings. ‘Waiver of consent’ was defined as situations where investigators are not required to inform or request permission from research subjects prior to participation.17 Emergency care research was excluded as such research involves critically unwell individuals who are unable to consent and/or involve time-critical emergency interventions, for which modified or delayed consent is typically employed (ie, a temporary waiver of consent followed by seeking consent at a later date once the patient is stable, or from an authorised representative or guardian). The ethical principles relevant to such situations are distinct from that of waiver of consent in non-emergency clinical research.
We included all available English-language ethical guidelines or statements on the use of waiver of consent for non-emergency clinical research. We searched bibliographic databases MEDLINE and EMBASE using the search terms: ‘research’ or ‘clinical research’ or ‘medical research’ or ‘health research’, and ‘consent’ or ‘informed consent’ or ‘ethics’ or ‘medical ethics’, and ‘waiver’ or ‘simplified’. We also conducted an extensive online search for ‘grey literature’ using the above terms on the rationale that documents relevant to this review may be contained within health research regulatory guidelines produced by government agencies and not published in peer-reviewed literature.
Patient and public involvement
Patients or the public were not involved in the design, conduct or reporting of this review.
International, regional and national guidelines on waiver of consent
Our search yielded 14 eligible reference documents. None were obtained through database searches of MEDLINE and EMBASE, and all were obtained from ‘grey literature’ sources. These statements included 1 international and 13 national/regional guidelines.
The International Ethical Guidelines for Health-related Research Involving Humans were first released in 1982 and have undergone several revisions, most recently in 2016.18 These international guidelines are a collaboration between the Council for International Organisations of Medical Sciences (CIOMS) and the WHO, and provide a global framework of ethical principles to guide the conduct of all health-related research involving human participants. Guideline 10 pertains to waiver of consent. In addition to the CIOMS international guideline, we identified 1 regional (European Union) and 12 national guidelines applicable in 39 countries (figure 1). These documents and their intended scope are detailed in table 1.

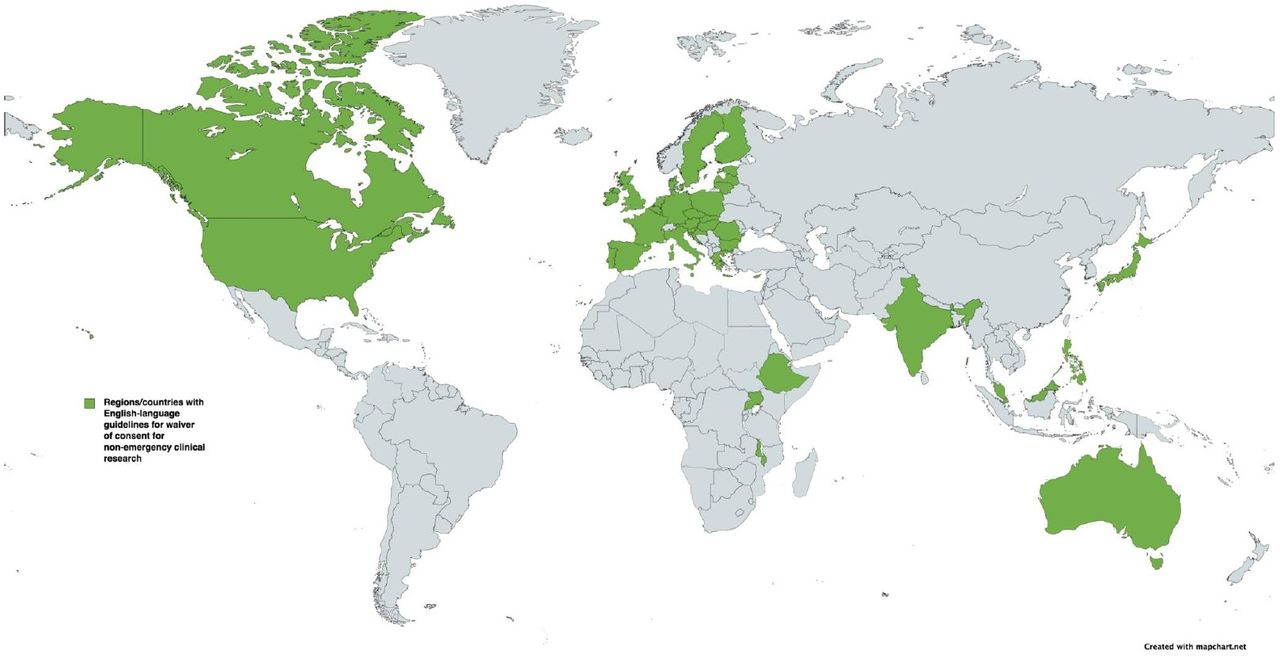{kind=link}
Countries with regional or national guidelines for ‘waiver of consent’ in non-emergency clinical research. Figure sourced from MapChart and modified with permission.
International, regional and national guidelines included for review within this paper with ‘waiver of consent’ guidelines for non-emergency clinical research
Within the CIOMS international guidelines, three essential criteria for using waiver of consent are described:
The research would not otherwise be feasible or practicable.
The research has important social value.
The research poses no more than minimal risk.
In addition to these criteria within the CIOMS guideline, other criteria were identified within national and regional guidelines for the use of waiver of consent and are summarised in table 2. Among these international, regional and national guidelines, we identified seven recurring principles which are discussed below.
Individual criteria for waiver of consent in non-emergency clinical research within international, regional and national guidelines
Review of individual principles
Would not otherwise be feasible or practicable
Research being ‘not otherwise feasible or practicable’ was a universal criterion within all guidelines examined.17–30 ‘Unfeasible’ and ‘impracticable’ are specifically defined in some,18 19 with a consensus towards a lack of scientific or logistical feasibility and onerousness that wholly jeopardises study conduct; mere inconvenience is not a sufficient justification.18 31 32
This criterion is the central theme underpinning waiver of consent research, with particular emphasis that informed consent should remain the standard where it is practicable or feasible to obtain consent, regardless of whether other criteria are met for waiver of consent.33 34 Specific descriptions for this criterion within the examined guidelines are outlined in table 3.
Detailed description of wording used for individual criteria contained within each international, regional and national guideline
Important social value and ‘risk-value’ assessment
The ‘important social value’ criterion is founded on beneficence ideals, with justification for carrying out the research being for the benefit of subjects, the community and society at large.35 36
This criterion is included, though variably worded, in several guidelines (table 3).18 19 22 25 In guidelines without a specific social value criterion for waiver of consent, social value is often stipulated as a broader requirement of all clinical research.17 21 24 26 27 29 30 Linked to this social value criterion is also the concept of balancing potential risks to participants against anticipated social value (‘risk-value’ assessment) which is expressed in some guidelines.19 21 30 35
No more than minimal risk
The requirement of ‘no more than minimal risk’ aims to uphold non-maleficence principles.31 ‘Minimal risk’ is specifically defined in 12 guidelines, with unifying concepts being that the anticipated probability and magnitude of harm is small. The concept is sometimes expressed as no or minimal additional risk from participation in the research relative to that of not participating (the background risk of the management of the condition in clinical care).17 19–27 29 30
The ‘no more than minimal risk’ criterion is contained within all guidelines examined (table 3).17–30 Four of the guidelines explicitly define ‘minimal risk’ as relating to both interventions/treatments and any associated tests/monitoring from conducting the research.20 26 28 30 Two of the guidelines explicitly refer to the testing of treatments currently available within clinical practice.20 30 The UK guidance document also comments on individual equipoise within cluster-determined interventions, stating that ‘healthcare professionals have the option of using an intervention other than the one assigned if they believe doing so is important for a particular patient’, relating to situations where clinician judgement deems neither clinical equipoise nor minimal risk apply.20
Protection of welfare and rights
The ‘protection of participant welfare’ criterion closely relates to ‘minimal risk’ and broader principles of non-maleficence.31 ‘Welfare’ is discussed in seven guidelines (table 3).17 19 20 22 24–26
The condition of a subject’s ‘rights’ not being adversely affected by the research is complex and raises the question of which rights are most relevant. Respect for individual autonomy may be thought to include the right to participate in research, choose between interventions and be informed throughout.35 However, Chalmers and Truog argue standard clinical care includes many low-risk decisions in which patients are neither involved in nor formally consented.11 37 Comparative effectiveness research and cluster randomised trials are frequently conducted in these usual care settings, and hence, it may be argued that participation need not involve any additional level of disclosure and consent.31 Acknowledging these contentions, the term ‘rights’ is detailed in the USA, UK, Philippines and Indian guidelines in conjunction with participant welfare20 24 26 38 and in the Malawian NHSRC guidelines which stipulate the ‘waiver is consistent with individual’s rights’.28 Furthermore, the UK draft guidance document requires that the ‘patient has not expressed a strong preference for any particular treatment’,20 and Australian NHMRC guidelines stipulate ‘there is no known or likely reason for thinking that participants would not have consented’,21 both of which pertain to maintenance of autonomy and individual rights.
Protection of privacy and confidentiality
Maintaining patient privacy and confidentiality is an expected feature of all clinical research.18 This is specifically detailed in two waiver of consent guidelines (table 3).21 22
Provision of information
Provision of pertinent information after research participation is founded on the ethical principle of respect,39 and it is detailed in nine guideline documents (table 3).19 21 24 25 27–30 38 Provision of such information after participation acknowledges that while these low-risk trials may have scientific justification for waiver of consent at the time of trial conduct, that patients remain the primary stakeholders and there is a responsibility to maintain public confidence and accessibility of the research process.
Several documents also discuss the provision of information on a community level prior to conducting the research, including Canadian Tri-Council guidelines which, in relation to population and public health research, state that ‘researchers should … seek community engagement prior to data collection’,19 Japanese MHLW guidelines state that measures be taken ‘to make announcement for the population to which the research subjects etc. belong’,25 and Ethiopian MoST guidelines state that ‘it is the responsibility of the investigator and the sponsor to publicise such a study to the community before commencement’.27
Legal and financial obligations
Reference to legal obligations, which closely relates to the principles of beneficence and non-maleficence, is discussed in two guidelines (table 3).21 30 Australian NHMRC guidelines also discuss financial obligations in reference to waiver of consent research, requiring that ‘the possibility of commercial exploitation of derivatives of the data or tissue will not deprive the participants of any financial benefits to which they would be entitled’.21
Implications of waiver of consent policy
The individual ethical principles represented within contemporary intentional, regional and national guidelines regarding criteria for the use of waiver of consent in low-risk clinical research indicate several unifying concordances of waiver of consent policy. Collation of these guidelines in this review demonstrates overarching inclusion of the three central criteria put forward in the CIOMS guidelines for waiver of consent; that the research would not otherwise be feasible, the research has important social value and the research is minimal risk. Additional criteria contained within national and regional guidelines aim to provide further protection of research participants while enabling low-risk routine care studies that could not otherwise be conducted.
The CIOMS guidelines and their adoption within national/regional guidelines provide a framework for understanding situations where waiver of consent may be appropriate in study design. Importantly, these guidelines are not designed to alleviate researcher inconveniences; rather, they aim to provide a model for conducting ethically founded research and improving health that would otherwise not occur if other, more traditional, consent models are required.34 37 In the face of uncertainty and variations in practice, requiring a higher threshold for conducting a formal evaluation of treatments in RCTs than for making clinical treatment decisions will result in a paucity of evidence-based practice with no gain for patient autonomy. An equally important consideration is that in situations of true clinical equipoise, where practice variability lacks consensus rather than reflecting good individualised decision-making, preventing research that could not otherwise be carried out without a waiver of consent will lead to continued treatment variability that poses at least the same risk to current and future patients as random treatment assignment.40
Historically, participants in RCTs across a variety of specialities are poorly representative of the real-world patient population for which the intervention is intended, and this reflects strict eligibility criteria, volunteer bias and likely other biases.4–7 These are typically trials of novel agents where the effects of the agents on human health, including risks, are largely unknown. By contrast, low-risk pragmatic RCTs and cluster randomised studies are conducted in situations where all patients already receive one of several potential treatments, the relative benefits and risks of which are uncertain but the decision on which treatment to use is not based on high-quality, direct evidence. These randomised studies can use broad inclusion criteria, reflect real-world clinical situations and generate evidence clearly generalisable to a wider patient population.41 Waiver of consent is of value in these situations where the research would not otherwise be feasible for methodological reasons (eg, interventions with cluster-level participation or cluster-level outcomes), where there is no or minimal risk (with ‘minimal’ referring to both the severity and the probability of risk), and where there is anticipated to be significant individual, community and/or societal value and real-world evidence from the research being conducted. Anchoring expectations of consent between clinical settings and corresponding real-world research settings and recognising the role that waiver of consent holds in conducting such low-risk research that would not otherwise be feasible is fundamental to broadening pragmatic clinical research and the evaluation of real-world healthcare interventions. Accordingly, clinical research involving waiver of consent is gaining increasing awareness and has been successfully and appropriately used in a number of clinical studies (table 4).
Examples of completed clinical research studies using waiver of informed consent
Ethics committees can provide valuable oversight in research conducted under a waiver of consent model. Well-resourced and well-informed expert ethics committees that are familiar with the ethical considerations for waiver of consent studies are best placed to protect the safety of participants. Oversight of data security frameworks is a key consideration in research conducted with a consent waiver. Human research ethics paradigms evolved to provide community oversight of and trust in medical research.42 Adherence to the highest standards in the conduct and oversight of waiver of consent studies is essential to maintaining public trust. Some statements explicitly suggest contemporaneous publication of waiver of consent approvals as a way of informing the community at large of research that involves individuals within that community.21 The role of guidance such as that presented in the current collation of waiver of consent statements is a step towards facilitating appropriate implementation of ethical principles and a common high standard in the conduct of waiver of consent research.
Strengths and limitations
International, regional and national ethical statements and guidelines on waiver of consent in clinical research historically have been presented in various online formats and have not been available in peer-reviewed published literature. This review collates a broad range of guidelines to provide researchers and ethics committees alike with a summary of principles that were deemed requirements for considering a waiver of consent model for any individual study. They support the design, oversight and conduct of collaborative multicentre international research that may warrant a waiver of consent model. However, there are important limitations to this review that should be considered. First, international, regional and national ethical statements and guidelines on waiver of consent in clinical research have been presented in various online formats and have not been available in peer-reviewed published literature. Although a detailed search was employed, it is possible some relevant documents may have been missed. Second, our inclusion criteria were restricted to English-language documents. Finally, ethical guidelines are subject to review and individual statements may be revised or modified over time. A dynamic online platform which collates real-time recommendations and guidance on human research ethical issues (including waiver of consent) would be worthy of consideration to assist researchers and ethics committees and enhance engagement from the broader community.
Conclusions
National, regional and international frameworks exist for the utilisation of waiver of consent in low-risk clinical research studies that closely resemble usual practice under the oversight of designated ethical bodies. This paper provides a summary of international approaches to waiver of consent research together with the level of consensus for the individual principles. These statements have a unifying basis consistent with the 2016 CIOMS guidelines with three overarching criteria: that the research would not otherwise be feasible, the research has important social value and the research is minimal risk. Additional criteria contained within some but not all national and regional guidelines aim to provide additional and appropriate protection to research participants. The guidelines collated in this review provide researchers with a clear and harmonised framework for conducting low-risk pragmatic clinical research that would not otherwise be feasible, through rigorous assessments of the relative effectiveness of variations in clinical practice, identification of optimal practices and, ultimately, leading to improved healthcare outcomes.
Ethics approval
Not applicable.
References
Footnotes
Collaborators The RESOLVE Study Global Team includes Grace Balicki, Abhinav Bassi, Sunita Bavanandan, Erika Dempsey, Carmel Hawley, Vivekanand Jha, James Kauffman, Behram Khan, Rathika Krishnasamy, Zuo Li, Vlado Perkovic, Jule Pinter, Patrick Rossignol, Michael Walsh, Christoph Wanner, David Wheeler.
Contributors MJ conceived the paper; AS wrote the first draft; and MJ, AS and BS all contributed to appraisal and reviewing. MJ is the guarantor of this work.
Funding The RESOLVE study is funded by competitive national grants: Australian National Health & Medical Research Council Project Grants (APP1127085, APP1187883), Canadian CIHR Innovative Clinical Trials Initiative (MYG-151209), United Kingdom National Institute for Health Research HTA Project (NIHR134660), German Research Foundation (DFG-Antrag PI 1689/1-1).
Map disclaimer The depiction of boundaries on this map does not imply the expression of any opinion whatsoever on the part of BMJ (or any member of its group) concerning the legal status of any country, territory, jurisdiction or area or of its authorities. This map is provided without any warranty of any kind, either express or implied.
Competing interests None declared.
Patient and public involvement statement Patients and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.