Article Text

Abstract
Objectives The aim of this study was to assess whether it was feasible to conduct a full trial comparing a tailored versus a standardised exercise programme for patients with shoulder subacromial pain.
Design Two-arm, patient-blinded and assessor-blinded, randomised controlled feasibility trial.
Methods Twenty-eight participants with shoulder subacromial pain were randomly allocated into one of two intervention groups—tailored or standardised exercise. Participants in the tailored exercise programme received exercises and manual therapy tailored to their scapular and shoulder movement impairments. Participants in the standardised exercise programme received progressive strengthening exercise. The primary outcome measures were (1) the participant recruitment rate; (2) the proportion of participants enrolled from the total number screened; (3) drop-out rates; and (4) adherence to the rehabilitation programme. Other outcome measures were: (5) pain levels; (6) Patient-Specific Functional Scale; (7) the Shoulder Pain and Disability Index; and (8) pain self-efficacy. We compared changes in pain and disability scores between groups using a repeated mixed-model analysis of variance. Since this is a feasibility study, we did not adjust alpha for multiple comparisons, and considered 75% CI as the probability threshold at 3-month follow-up. Health-related quality of life was assessed using the Short-Form 12 and quality-adjusted life years (QALYs) were estimated.
Results The recruitment rate was 3 participants per month, the proportion of participants enrolled was 23%, the drop-out rate was 14% and the overall adherence to the rehabilitation programme was 85%. No between-group differences were found for most outcome measures. Adverse events (n=2, only in the tailored group) were minor in nature and included skin injury or pain following taping.
Conclusions Our feasibility trial showed that additional strategies are required for improving recruitment, enrolment and minimising drop-out of participants into the trial and making it feasible to conduct a full trial.
Trial registration number ANZCTR: 12617001405303.
- clinical trials
- shoulder
- elbow & shoulder
- rehabilitation medicine
- pain management
Data availability statement
Data are available upon reasonable request. The datasets generated during the study will be available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The protocols used for both intervention arms had detailed information about how to progress each of the included exercises over the intervention period.
Clinicians received training sessions to familiarise themselves with the protocol and the trial only started after clinicians received the training and considered themselves familiarised with interventions from both arms.
Efficiency of recruitment and enrolment, participant adherence and retention were exposed as limitations of the study design; however, most participants did adhere to the rehabilitation programme, and the drop-out rate was within a priori bounds.
Session duration is not representative of current practice in New Zealand, limiting generalisability; however, current practices should not restrain research from testing new intervention practices that may deliver better outcomes for patients with shoulder disorders.
Introduction
Shoulder pain is the third most common musculoskeletal problem, with a 1-year prevalence of 18.1%.1 This high prevalence in combination with the significant disability caused by shoulder pain results in high burden—the average annual cost of shoulder subacromial pain has been estimated at $4139 per patient, in Sweden,2 and in New Zealand costs for shoulder injuries totalled NZ$14 million/year in on average from 2005 to 2013.3 Shoulder subacromial pain is defined as pain at the top and lateral part of the shoulder joint, may spread to the neck and elbow and is worsened by overhead activity.4 It has a slow recovery,5 with only 50% of new episodes presenting full recovery within 6 months.6
Best evidence recommends exercise therapy be prescribed for patients with shoulder subacromial pain.7 8 However, the strength of evidence supporting this recommendation is limited and findings from two large trials found exercise therapy did not provide additional benefit over usual care.9 10 On the other hand, a recent systematic review and network meta-analysis suggested that, among other interventions, exercise and manual therapy are likely to be effective in the short term for pain and function outcomes.11 Currently, it is uncertain: (1) if exercise therapy is more effective than placebo; (2) which form of exercise therapy is likely to be more effective; (3) whether exercise combined with manual therapy is likely to be more effective than exercise alone.
There is conflicting evidence in the literature regarding effectiveness of exercise therapy when compared with placebo (ie, detuned ultrasound or detuned laser therapy). One trial found exercise and manual therapy are no different to detuned ultrasound,12 while another trial found exercise therapy to be more effective than detuned laser therapy.13 The last Cochrane Review recommended future trials to compare exercise interventions with placebo.14
With regards to the type of exercise, one large trial reported specific exercise programme (targeting rotator cuff and scapular muscles) to be more effective than a generic strengthening exercise programme.15 However, findings from a systematic reviews suggest limited evidence regarding the effectiveness of specific resistive exercise when compared with general strengthening exercise.16 Two recent reviews suggested future trials to compare different types, dose or duration of exercise therapy regimens.16 17
The role of manual therapy in the management of patients with shoulder subacromial pain is unclear and debated in the literature.17 There are conflicting recommendations from previous trials and reviews in the topic.11 14 17 18 Evidence from trials on other musculoskeletal disorders suggests that including manual therapy led to better clinical outcomes when compared with corticosteroid injection or wait-and-see in the management of other musculoskeletal disorders (eg, tennis elbow),19 or usual care when managing patients with hip or knee osteoarthritis.20 A recent systematic review and network meta-analyses suggested exercise and manual therapy are likely to have small to moderate treatment effects on patients with shoulder subacromial pain, but the level of certainty was low.11
Together, findings from those previous trials and systematic reviews suggest it is unclear whether exercise therapy (when combined or not with manual therapy) is effective for managing patients with shoulder subacromial pain in comparison with placebo or usual care. It is also unclear which form of exercise therapy interventions is more likely to be effective for improving pain and function in those patients. The aim of our full study is to assess the clinical efficacy and cost efficacy of a tailored exercise programme versus a standardised exercise programme versus usual care for the treatment of shoulder subacromial pain.
Prior to conducting a fully powered randomised controlled trial (RCT), we conducted a feasibility trial to assess: (1) the participant recruitment rate; (2) the proportion of participants enrolled from the total number screened; (3) adherence to the exercise programmes; (4) drop-out rates; (5) preliminary estimates of adverse events; (6) preliminary estimates of intervention effects in order to inform the sample size of the fully-powered RCT; and (7) the feasibility of collecting costs-related data within the trial.
Methods
Trial design
The management of subacromial disorders of the shoulder trial is a two-arm, patient-blinded and assessor-blinded, feasibility RCT. Participants were randomly allocated into one of two intervention groups: a standardised or a tailored exercise programme.
We followed the Consolidated Standards of Reporting Trials 2010 statement: extension to randomised pilot and feasibility trials.21 In addition, we followed the Template for Intervention Description and Replication checklist and guide.22 The study protocol was prospectively registered with the Australian and New Zealand Clinical Trials Registry (12617001405303) and published.23
Participants
We recruited participants with shoulder subacromial pain, aged from 18 to 65 years old, from within the Dunedin area (New Zealand) through newspaper advertisements.
Participants were screened by a musculoskeletal physiotherapist, following the British Elbow and Shoulder Society (BESS) guidelines.4 Given the challenges in diagnosing patients with shoulder pain and the low sensitivity of most clinical tests for the shoulder disorders,24 we widened the criteria proposed by BESS and added resisted lateral rotation and shoulder abduction tests.25 The BESS guidelines screen for red flags (eg, tumour, unreduced dislocation, acute rotator cuff tear, infection), shoulder pain with cervical spine origin, shoulder instability, acromioclavicular joint disease or adhesive capsulitis.4
Participants were included if they presented a positive finding on one of the following tests: (1) painful arc movement during shoulder flexion or abduction; (2) Jobe’s test4; or (3) pain on resisted lateral rotation or abduction.25 We excluded participants with a history of shoulder dislocation, shoulder subluxation, shoulder surgery and cervical surgery within the last 6 months,26 participants with any kind of symptoms of systematic inflammation or disease, signs of paraesthesia in the upper extremities, hemiplegic shoulder pain, frozen shoulder or positive clinical signs of full thickness rotator cuff tear.27
All participants provided written consent prior to taking part in the study.
Interventions
Participants in both groups received 16 individual, face-to-face sessions, each lasting for approximately 60 min, over an 8-week period. Details of interventions can be found in the published protocol.23 Participants were encouraged to not undertake any other treatment during the trial, but could do so, should they wish to pursue that. We asked participants to report any concurrent treatment during the trial.
Participants performed eight exercises per session, plus three stretches (control group) or up to three manual therapy techniques (tailored group). To enhance the internal validity of the trial, the number of exercises and duration of sessions were planned to be equivalent. The intensity of strengthening exercises was monitored using a modified Borg Scale.28 The Borg Scale is valid tool for measuring exertion during resistance training.29 30
Tailored exercise programme: participants allocated to the tailored exercise programme received exercises focusing on restoring normal movement pattern and the dynamic stability of the scapulothoracic and glenohumeral joints,31 32 in addition to manual therapy techniques for restoring shoulder and scapular movement33 and progressive resistance training of impaired muscles.32 34 Theoretically, this intervention should lead to better clinical outcomes given it targets specific neuromuscular and join impairments presented by the patient.
Standardised exercise programme: participants allocated to this group received progressive resistance training for all scapular and shoulder muscles and a stretching exercise programme.35 This intervention focused on restoring muscle flexibility and strength.
Primary outcome measures
The primary outcome measures were: (1) the participant recruitment rate, measured as number of participants enrolled per month; (2) the proportion of participants enrolled from the total number screened, with reasons for exclusion; (3) drop-out rates, expressed as a percentage of the total number of participants enrolled; and (4) adherence to the exercise programme, measured as number of sessions attended as a percentage of the total number of planned sessions.
Other outcome measures
Other outcomes were collected via face-to-face interviews. When selecting outcome measures to use for this feasibility trial, we considered the patient-reported outcome measures intended as the primary and secondary outcomes to be used in the main trial. Hence, the outcome measures were:
Pain intensity (at rest, during arm movement and average pain during the last 7 days) measured by a numeric pain scale.36 The numeric pain scale is a reliable and responsive tool when used with patients with shoulder pain.37 The minimal clinically important difference (MCID) for the 10-point numeric pain scale in patients with shoulder pain is 1.1 points.37 High scores represent worse outcomes.
The Patient-Specific Functional Scale (PSFS). The PSFS measures disability and is a valid, reliable and responsive tool for assessing patients with shoulder pain.38 The MCID for the PSFS is 1.3 (for small changes), 2.3 (medium changes) and 2.7 (large changes) in patients with a range of musculoskeletal disorders.39 Low scores represent worse outcomes.
The Shoulder Pain and Disability Index (SPADI) total score (including the pain and disability subscales).40 The SPADI presents acceptable construct validity and responsiveness in patients with shoulder pain.41 According to a systematic review, the MCID for the SPADI total score ranges from 8 to 13.42 High scores represent worse outcomes.
The pain self-efficacy questionnaire.43 The pain self-efficacy questionnaire is an established and commonly used tool for assessing self-efficacy in individuals with pain.44 The MCID for the pain self-efficacy questionnaire is 9 points for patients with low back pain.45 Low scores indicate low levels of self-efficacy when dealing with pain.
We assessed safety by recording all adverse events, both related and unrelated to interventions, in each group. The literature suggests adverse events to exercise therapy might be common, but not serious.46 Potential adverse reactions to interventions may include increased pain around the shoulder joint. The physiotherapist recorded any adverse reactions to interventions, including duration and severity of adverse reaction to treatment, and how the adverse reaction was managed. We included in the report the total number of participants who reported adverse events, relatedness to interventions, and the duration and severity of the adverse reactions. In the small sample of this feasibility trial, we did not expect to observe a representative number of adverse events, so did not undertake statistical comparisons.
Economic outcomes
Health-related quality of life was assessed using the Short-Form 12 (SF-12v2) questionnaire.47 To allow the calculation of health utility values for the economic evaluation the SF-12v2 was converted to a six-dimensional health state classification (SF-6D).48 Health utility is a preference-based measure of overall health-related quality of life, on a scale from 0 (equivalent to death) to 1 (full health). Quality-adjusted life years (QALYs) were estimated for each participant by calculating the area under the curve (the product of utility values by time) from baseline to 12-week follow-up. We calculated the mean QALYs for each group and adjusted for baseline utility scores to minimise any bias due to chance of baseline imbalance between the groups.
We adapted the Otago Cost and Consequences Questionnaire (OCC-Q) to shoulder disorders and used the adapted questionnaire to capture healthcare use and other non-healthcare costs (eg, time off work).49 The OCC-Q is a validated patient-administered questionnaire developed for osteoarthritis that has demonstrated accuracy and agreement with administrative databases in the New Zealand healthcare system.49 The OCC-Q was administered at baseline and 12-week time points. Costs are expressed as 2019 NZ dollars, exclusive of Goods and Services Tax.
Sample size
Given this is a feasibility trial, we did not design it to assess the efficacy of the experimental intervention.50 51 Whitehead et al52 recommend the sample size of a feasibility study should be estimated based on the expected range for the effect size, the power and alpha (both established a priori), and the total number of arms of treatment planned for the full trial.52
Whitehead et al52 estimated the sample size based on standardised differences of different magnitudes (ie, extra small, small, medium and large). To estimate sample size, we used the SPADI as the presumed primary outcome measure for the full trial and assumed a minimum clinically important difference of 8 points,53 with an SD of 24 points.53 This represents a standardised effect size of 0.3. We considered a full trial with power of 80%, two-tailed between-group comparison, and alpha at 0.05. Therefore, the minimum sample size for this feasibility RCT is 10 participants per arm of treatment, assuming a medium effect size.52 Assuming a 20% loss to follow-up,54 we aimed for a minimum sample size of 25 participants.
Randomisation
Sequence generation, allocation concealment, implementation
Participants were allocated (1:1 ratio) into one of the intervention groups (ie, tailored physiotherapy or standardised physiotherapy) through blocked randomisation (with blocks of 4). The randomisation schedule was computer-generated by a research administrator not involved with delivering the interventions, and concealed in numbered, sealed and opaque envelopes. A research administrator provided the envelope to the clinician delivering the interventions.
Blinding
Participants and outcome assessors were blinded to group allocation. Clinicians delivering the interventions were not blinded to group allocations due to nature of interventions.
Time points
Outcome measures were recorded at baseline and at the 4th, 8th and 12th weeks after baseline.
Statistical analysis
We used descriptive statistics analyses for presenting: (1) recruitment rates; (2) proportion of participants enrolled from the total number screened; (3) drop-out rates; (4) adherence to the exercise programme; and (5) adverse events and for reporting economic outcomes. The primary and secondary analyses were intention-to-treat and involved all patients who were randomly assigned. All statistical analyses were conducted using R.55
We used linear mixed-effect models to obtain estimates of treatment effects. We conducted within-group and between-group comparisons using an independent linear mixed-effect model for each outcome measure (ie, numeric pain rating scale, PSFS, SPADI pain score, SPADI disability score, SPADI total score, and pain self-efficacy). This feasibility trial was not powered to detect superiority; however, we assessed the magnitude of mean treatment effects for pain and disability in relation to clinically important changes. This was done for informing the choice of primary outcome measure to be used in the main trial and thus informing the sample size calculation for the main trial.56 When running linear mixed-effect models, we estimated marginal means and their respective 75% CIs. For that reason, we did not adjust alpha for multiple comparisons. This statistical approach is considered appropriate for feasibility or exploratory studies.57
When conducting within-group comparisons, group allocation (tailored and standardised exercise groups) and ‘time-point’ (baseline, 4th, 8th week and 12th week) were considered as fixed-effects. Participants were considered as random effects. Post-hoc analyses were conducted for comparing changes in scores between ‘baseline vs 4 weeks’, ‘baseline vs 8 weeks’ and ‘baseline vs 12 weeks’.
When conducting between-group comparisons, group allocation (tailored and standardised exercise groups) and time-point (4th, 8th week and 12th week) were considered as fixed-effects. Participants were considered as random effects. Baseline measurements were considered as covariates. Post-hoc analyses were conducted for comparing scores between groups at each time point (ie, 4, 8 and 12 weeks).
To help inform whether it is worthwhile conducting the full trial, it is recommended that preliminary between-group comparisons be performed at the feasibility trial stage.58 59 For that, CI ranges other than 95% are recommended when assessing between-group differences from feasibility trials (eg, 75% CI in addition to the mean difference estimate).58 For the purposes of this study, we considered 75% CI as the probability threshold for between-group analyses.58 Such information will be considered when assessing whether to conduct the full trial.58 59
Missing data
Linear mixed-effect models can handle missing data. For descriptive analysis, in case of missing data, we explored pattern of missingness using the ‘mi’ package in R.60 After running such analysis, we accepted that data were missing at random and performed multiple imputation by chained equations using the ‘mice’ package.61
Additional analysis
When running the mixed-effect models, we found residuals presented small deviations from the normal distribution. In those cases, it is recommended to conduct robust mixed-effect models and report estimates from both models (ie, standard and robust mixed-effect models).62 We implemented the robust mixed-effect models using the ‘rlmer’ function from WRS 2 package.62 The robust models used the same dataset as the standard mixed-effect models (described above) and yielded similar estimates of treatment effects to those obtained with standard mixed-effect models. For that reason, we report in the main text the estimate effects obtained through the mixed-effect models and reported the estimate effects obtained through the robust mixed-effect models in the online supplemental material 1.
Supplemental material
Results
Recruitment and flow of participants
The recruitment flow and randomisation process are presented in figure 1. The trial started recruiting on 19 January 2018 and completed recruitment on the 23 October 2018. The trial ended after recruiting the minimum number of participants as per sample size calculations.

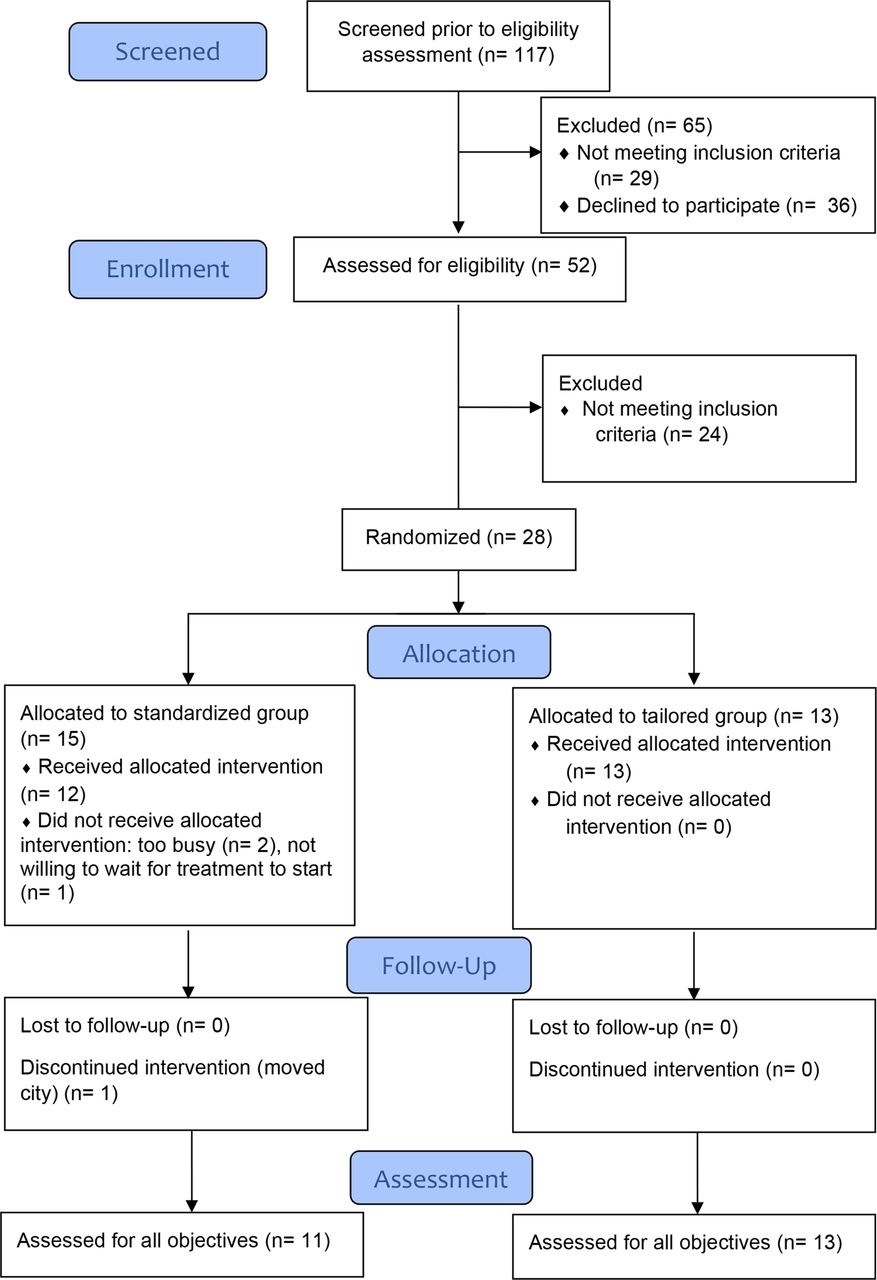{kind=link}
Flow of participants in the trial.
A total of 117 individuals showed interest in taking part in the study and completed telephone screening; 51 were excluded at that screening stage. The main reasons for exclusion were inability to commit to the study, no response after receiving the information sheet or not meeting the inclusion criteria.
Fifty-three participants were physically screened, with 24 participants excluded following physical screening. Reasons for being excluded included (with some participants meeting more than one exclusion criteria): not presenting positive tests to physical examination of the shoulder (n=12), symptoms caused by neck disorder (n=7), history of subluxation (n=1), frozen shoulder (n=2), acromioclavicular joint involvement (n=4), inflammatory disease (n=3). Following physical screening, 28 participants were eligible for randomisation.
Participants’ characteristics
The demographics and clinical characteristics of participants are presented in table 1.
Baseline characteristics of 28 participants
Outcomes and estimation
Primary outcome measures
Findings for primary outcome measures are presented in table 2. The proportion of participants enrolled from the number of participants screened was 23%. Participant recruitment rate (number of participants recruited per month of active recruitment) was 3. The drop-out was 14% for all participants enrolled in the trial. Four participants allocated to the standardised intervention dropped out. One participant dropped out of the study due to relocation to another city. The other three participants withdrew before initiating physiotherapy intervention, the reasons for dropping out were: not able to commit to the study (n=2) and not wishing to wait for the start of interventions (n=1). All participants allocated to the tailored exercise programme completed the trial. The adherence to the exercise programme was 85% for all participants combined, with 73% for participants allocated to the standardised group and 100% for participants in the tailored group.
Descriptive statistics for primary outcome measures
Other outcome measures
The descriptive mean scores for pain, disability and pain self-efficacy are presented in table 3. The within-group changes are presented in the online supplemental material 2. The estimated marginal mean for between-group differences and their respective 75% CIs are presented in the online supplemental material 2. The estimated marginal means obtained with the standard and robust mixed-effect analyses are presented in the online supplemental material.
Supplemental material
Participants’ scores for pain, disability and function at each time point (mean and SD)
Economic outcomes
The total costs regarding visits to healthcare practitioner, healthcare tests or treatment or pain medications at 12 weeks follow-up and mean QALYs are presented in table 4.
Total costs (in 2019 NZ$) and health outcomes at 12-week follow-up
Harms
All adverse events were considered minor events. A total of two adverse reactions were reported, all by participants allocated to the tailored group (table 5). Adverse reactions skin injury following taping of the shoulder and increase in shoulder pain following taping of the shoulder.
Adverse reactions reported by participants following treatment
Discussion
This trial assessed the feasibility of conducting a full trial that will compare two forms of exercise therapy for patients with shoulder subacromial pain (one tailored and one standardised exercise programme). Overall, our findings suggest it is feasible to conduct the full trial given that most participants adhered to the exercise programme, and the drop-out rate was within a priori bounds. However, prior to conducting the full trial, few amendments to the design are required.
We identified limitations that must be addressed when designing the full trial. Our recruitment rate was lower than previous full trials8 63 but similar to a previous feasibility trial.64 Our ability to enrol participants into the trial during the 9-month period of recruitment was limited by the number of clinicians involved with the study. That impacted on recruitment rate and that can be addressed in the future trial by having a multicentre design.
For the present study, the clinic responsible for delivering the interventions limited the number of participants that could be treated to a maximum of 10 at any given time. That impacted on flow of participants in the trial and prevented us from continuously enrolling participants. For that reason, we had to recruit participants in three stages. Some participants opted to drop-out after being screened for eligibility and notified that there would be a waiting period for interventions to start.
We recruited participants through a local newspaper. This may explain why most of our participants presented mild to moderate pain intensity. Participants in our study presented lower pain or function scores compared with those from previous full trials.8 10 65 66 For the full trial, we plan to adopt a multimodal recruitment strategy, including general practice clinics, social media and waiting list from local hospitals. Such strategy may help to optimise recruitment rate and recruit patients with higher levels of shoulder pain or disability.
Our sample presented similar scores for pain and slightly lower scores for disability at baseline compared with a large trial with participants with shoulder subacromial pain.12 Participants in both groups were exposed to active interventions and presented similar changes in pain and function scores over time. The magnitude of changes in pain scores at 12 weeks was greater than those reported by participants exposed to exercise therapy or placebo intervention reported by one large trial.12 Feasibility and pilot trials are notorious for their imprecise estimates of treatment, given their small sample sizes.67 For the full trial, we will include a control arm (eg, an inactive control such as detuned therapeutic ultrasound or laser, or usual care) to be able to estimate the effect of standardised or tailored interventions on clinical outcomes. This strategy has been successfully used before.
When designing the future trial, we will consider a multicentre design to ensure the minimum sample size required for the full trial is met. Multicentre trials tend to provide treatment effects that are smaller when compared with single-centre trials. In addition, multicentre trials tend to be more pragmatic than smaller trials. It is suggested that the estimate treatment effect observed in a multicentre trial is closer to those we would observe in clinical practice.68–70 For those reasons, multicentre trial is more relevant and useful for clinicians, patients and policy-makers than single centre trials.
Our findings helped to identify the primary outcome measures to use in the full trial. According to a recent Delphi study, trials on shoulder disorders should assess the following domains: pain, physical functioning, global assessment of treatment success and health-related quality of life.71 Based on our findings, pain during arm elevation presented the largest changes from baseline to 12-week follow-up for both groups. Recently, it has been recommended that movement-evoked pain should be used for assessing musculoskeletal pain.72 Our findings also suggested important within-group changes for PSFS and SPADI scores and either of those outcome measures could be used in the full trial. The advantage of PSFS is that it assesses tasks that are especially relevant for a given participant,40 while SPADI suffers the limitation of fixed-item instruments, where some items in the questionnaire may not be relevant to a given participant.40 Based on that, PSFS should be considered as a primary outcome measure in the full trial.
In this feasibility study, we did not assess the ‘global assessment of treatment success’ and the future trial should include an outcome measure assessing that construct. We assessed health-related quality of life using the SF-12v2 questionnaire and that should be included in the full trial. When designing the final trial, we will follow the most current recommendations and future work by the Outcome Measures in Rheumatology (OMERACT) Shoulder Working Group.71 73
Strengths and limitations
The trial design had some notable strengths. The protocols used for both intervention arms had detailed information about how to progress with exercises over the intervention period. Clinicians received training sessions to familiarise themselves with the protocol and the trial only started after clinicians received the training and considered themselves familiarised with interventions from both arms. We adopted clinical outcomes that are recommended for trials recruiting patients with shoulder disorders.73 Despite the longer duration of interventions, compared with current practice in New Zealand and other countries, participants adhered to both exercise programmes. The number of participants dropping out was low. Compared to the tailored group, the standardised exercise group, had a larger number of participants (n=3) dropping out after enrolment . The drop-out occurred before starting interventions. Findings from the full trial will help to identify whether a tailored or standardised exercise programme is more effective than a control intervention, reducing the socioeconomic burden of shoulder subacromial pain.
One criticism of our design is the duration of the interventions (ie, sessions lasting for 40–60 min), which is not representative of current practice in New Zealand. On the other hand, findings from one trial suggested higher dosage of exercise therapy led to better clinical outcomes.74 In addition, current practices should not restrain research from testing new interventions that may deliver better care for patients with shoulder disorders. While our trial will not compare different exercise therapy dosages, it will add valuable information regarding the effect of different forms of exercise therapy delivered at equivalent dosage. In addition, as per our protocol, our nested process evaluation study was conducted parallel to this feasibility trial. Findings from this process evaluation study will provide more detailed information regarding the participants’ and clinicians’ perceptions of the interventions tested in this feasibility trial. We have conducted a focus group with clinicians and individual interviews with patients who took part in the study to assess their perceptions about the interventions received. These findings will be prepared for publication as separate manuscripts. The information from the current study and the nested process evaluation study will be used for improving the design of the full trial.
Conclusions
Our feasibility trial showed that additional strategies are required for improving recruitment, enrolment and minimising drop-out of participants into the trial. By adopting additional strategies and addressing some of the limitations identified through this feasibility study, it is likely feasible to conduct a full trial assessing the efficacy of a tailored exercise programme.
Data availability statement
Data are available upon reasonable request. The datasets generated during the study will be available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the University of Otago Ethics Committee [H17/080]. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank Mr Andrew Grey for statistical advice and the financial support from the Health Research Council New Zealand. We also thank Physiotec® for permitting us to use exercise images from their exercise database (https://www.physiotec.ca/index.php).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @danielcr
Contributors DCR and ZJT conceived the research question. DCR was responsible for the design of the trial and is the guarantor. ZJT and GS contributed to the design of interventions. JHA provided guidance on the design the trial and economic analysis. RW provided guidance on economic analysis. DCR led efforts for securing funding, with the contributions from ZJT, GS and JHA. All authors revised and approved the protocol for the study. All authors revised the manuscript for important content and approved the final version.
Funding This work was supported by Health Research Council New Zealand (Grant number: 17/536). The trial sponsor is the University of Otago. The trial started in August 2017, and is funded until July 2019. This research was conducted during tenure of The Sir Charles Hercus Health Research Fellowship of the Health Research Council of New Zealand (Grant number: 18/111) awarded to DCR.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.