Article Text

Abstract
Ehlers-Danlos syndromes (EDS) are a group of connective tissue disorders associated with skin, ligament, blood vessel and organ abnormalities. Skin hyperextensibility, joint hypermobility and widened atrophic scars are characteristic of classical EDS. Vascular complications, though rare in classical EDS, can be life-threatening, and this necessitates one to look for vascular associations in non-vascular, such as classical, forms of EDS due to the heterogeneity of the syndrome. Reports of vascular complications in classical EDS are often limited to haematomas being the most frequent manifestation. This case report discusses an elderly patient with genetically confirmed classical EDS who suffered from a series of pulmonary and vascular complications, including recurrent spontaneous haemopneumothorax, aortic dissection and eventual mesenteric haemorrhage, which resulted in his death. Identifying clinical red flags is crucial to predict such future catastrophic vascular events and guide appropriate counselling and management strategies for individuals with classical EDS.
- Connective tissue disease
- Musculoskeletal syndromes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Ehlers-Danlos syndromes (EDS) are a heterogeneous group of connective tissue disorders often grouped with Marfan and Loeys-Dietz syndromes. These syndromes have variable abnormalities of the skin, ligaments, blood vessels and viscera. The prevalence of EDS is estimated at 1 in 5000 births, lacking ethnic, racial or geographical predisposition.1 The diverse genetic and molecular changes found in EDS result in poorly described subtypes and overlapping phenotypes. Despite this diversity, skin hyperextensibility, joint hypermobility and widened atrophic scars remain the key diagnostic features in classical EDS, with minor manifestations such as bruising or bleeding diathesis.2 In contrast, vascular EDS predominantly presents with thin, translucent skin and a tendency for the rupture of hollow organs and arteries due to generalised vascular fragility, resulting in the worst prognosis among EDS subtypes.2 We present a case of an elderly man with genetically confirmed, classical EDS, presenting with cumulative and devastating vascular complications throughout his life, resulting in his mortality.
Case presentation
Our patient was initially evaluated for possible EDS in his 20s in the context of recurrent primary pneumothorax. His history was significant for increased skin laxity, most prominent at the neck, thigh and forearm, which worsened with age. He had a history of easy bruising post-contact sports with thigh and forearm haematomas. Skeletal deformities included a moderate pectus excavatum which progressed with age. However, there were no spine deformities, nor any family history of connective tissue disorders. He was diagnosed with classical EDS (type II) at the age of 26 based on the clinical findings of increased skin hyperextensibility, atrophic scarring, mildly increased range of motion of joints and a history of superficial skin bruising with no Marfanoid ectasia. His developmental milestones were age-appropriate, and there were no major health concerns until adulthood. His initial presentations to the hospital were due to recurrent spontaneous pneumothorax, primarily of the left lung, necessitating intercostal drains.
However, at the age of 30, he experienced his first vascular complication, which was a massive spontaneous haemothorax of the left lung. This was initially treated by intercostal drain followed by definitive management with talc pleurodesis. He did have small recurrent right-sided pneumothoraxes in his late 30s and 40s, but no documented vascular events were noted during this period.
In his 50s, there were episodes of new atraumatic bruising in the scalp and knee, which however did not require hospitalisation. He had a recurrence of pneumothorax secondary to bullous emphysema resulting from his 30-pack-year smoking. Though this was considered a secondary pneumothorax, his EDS would have been a factor due to the challenged pleural interface. This prompted molecular testing, revealing a COL5A2 mutation in exon 29, c.1977 G>A, which is genetically linked to classical EDS. Evaluation for Marfan, Loeys-Dietz and EDS IV syndrome was negative. Following this, he reformed his smoking habit for the past 30 years.
In his 70s, he continued to have significant traumatic limb bruising from falls and frailty. The most notable event was a large posterior scalp haematoma secondary to forehead trauma, which was suspected to be a ‘contrecoup’ vascular scalp event. Recurrent falls with bleeding compromised his independence, and he was transitioned into residential care living.
His life-threatening vascular complications from EDS were first experienced in his 70s—five decades from initial diagnosis, during an episode of chest pain where he was diagnosed with a spontaneous Stanford B aortic dissection. A CT aortography revealed extensive intramural haematoma of the thoracoabdominal aorta commencing distally to the left subclavian artery and extending to the aortic bifurcation. This occurred in the absence of pre-existing hypertension or aortic aneurysm. He was managed medically with good outcomes.
Subsequently, 12 months later, he presented with paroxysmal palpitations and chest pains for 1 week, with no preceding falls or trauma. On examination, he was breathless with a blood pressure of 130/60 mm Hg, an irregular heart rate of 140–150 /min and a saturation of 94% while on his baseline domiciliary 3 L/min oxygen for his progressive emphysema. Clinical examination revealed bilateral fine crackles with an end-expiratory wheeze and pitting oedema to both his ankles, confirming a congestive heart failure precipitated by new-onset atrial fibrillation. Later during the night, he complained of acute abdominal pain, with repeat examination revealing generalised abdominal guarding and rigidity raising concerns of an acute thromboembolic ischaemic bowel secondary to atrial fibrillation.
Investigations
Blood investigations revealed haemoglobin of 123 g/L (135–175 g/L), white cell count 14.6 (4–11 × 109 /L), platelet 370 (150–450 × 109 /L), C-reactive protein 50 (<10 mg/L), troponin 55 (<16 ng/L), creatinine 82 (60–110 umol/L) and NT-pro BNP 21029 (<124 ng/L). Chest X-ray showed cardiomegaly with emphysematous changes and chronic lung bases atelectasis, with no consolidation or effusion. An emergency CT chest aortogram did not show evidence of dissection or dissection flap.
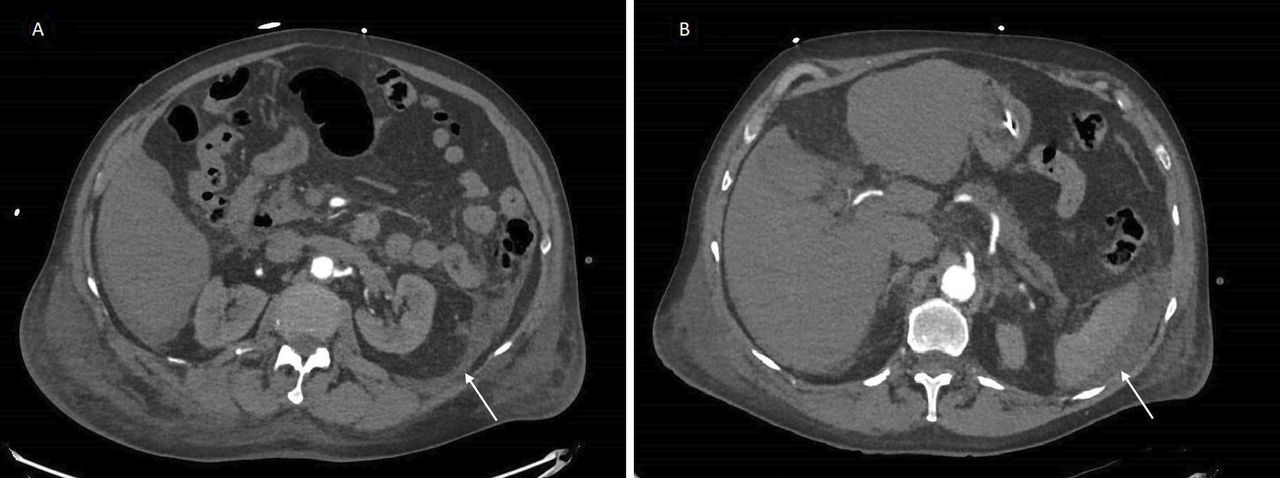
A venous blood gas performed during the episode of abdominal pain revealed pH 7.25 (7.35–7.45), pCO2 46 (35–45 mm Hg), bicarbonate 20 (22–26 mmol/L), lactate 11 (<2 mmol/L), consistent with mixed metabolic/respiratory acidosis and severe hyperlactatemia. Haemoglobin initially remained stable at 125 g/L. An urgent CT angiogram of his abdomen and pelvis revealed interval development of peritoneal fluid surrounding the descending colon and spleen, with free pelvic fluid (figure 1). The radiologist opined the presence of significant new intra-abdominal fluid represented a very high likelihood of mesenteric haemorrhage given the hypovolemic shock.

Axial CT of the abdomen and pelvis showing peritoneal fluid (white arrows) tracking down from the descending colon (A) to the left retroperitoneum and spleen (B).
Differential diagnosis
His presenting symptoms of paroxysmal palpitations and chest pains with peripheral oedema suggested an arrhythmia with right ventricular failure. This was evident as new atrial fibrillation was noted on ECG and chest X-ray (figure 2), demonstrating cardiomegaly allowing for AP (anterior-posterior) projection and markedly elevated NT-pro BNP. There were no radiographic features of pulmonary oedema or effusions to suggest a haemothorax given his propensity for it. The degree of cardiomegaly on X-ray was not typical for a pericardial haemorrhage or cardiac tamponade. Echocardiography, an important diagnostic and monitoring tool for heart failure, could not be completed given the patient’s acute presentation with rapid deterioration. There was persistent elevation of the right hemidiaphragm with right basal compressive atelectasis, consistent with underlying diaphragmatic eventrations secondary to tissue laxity in EDS, with no evidence of the rarer diaphragmatic rupture.

{kind=link}
{kind=link}
Chest X-ray (anterior-posterior sitting view) revealing cardiomegaly with right diaphragmatic eventration and basal atelectasis.
The initial considerations for our patient were ischaemic bowel given his vascular risk factors and new atrial fibrillation or recurrence of aortic dissection. Of note, the CT angiogram of his abdomen and pelvis showed no definite obstruction or dissection within the abdominal aorta, nor in the small peritoneal mesenteric vessels. Furthermore, there was no thumbprinting, wall thickening or colonic wall gas to suggest gut ischaemia or evidence of bowel obstruction. Given his tissue fragility, an intestinal rupture could account for his acute abdomen, biochemical derangements and hyperlactatemia; however, there were no visceral abnormalities or perforations on CT imaging. Another rare possible complication of hypermobility type EDS is visceroptosis; however, CT imaging did not reveal this. Investigative barium and transit studies were not considered given the clear evidence pointing to mesenteric haemorrhage.
Treatment
Early in his presentation, the cardiology team led the initial management of his atrial fibrillation and heart failure, focusing on rate control with metoprolol and digoxin, diuresis with frusemide and anticoagulation with apixaban in accordance with atrial fibrillation protocol and CHA₂DS₂-VASc score of 3 (age, heart failure, vascular disease/aortic dissection). His moderate bleeding risk with HAS-BLED score of 2 did not outweigh the benefit of anticoagulation. Apixaban was chosen for its established evidence and safety profile for stroke prophylaxis in non-valvular atrial fibrillation compared with other direct oral anticoagulants or warfarin.3 An alternative would be enoxaparin in situations with increased bleeding risk. However, our patient was given a single dose of apixaban prior to his development of an acute abdomen, and this was subsequently ceased due to the mesenteric haemorrhage.
Subsequent multidisciplinary management involved surgical and intensive care unit consults to treat his mesenteric haemorrhage. Acute surgical reviews deemed he was inoperable due to his comorbidities posing a high surgical risk. Interventional radiology was also consulted for arterial embolisation; however, an absence of arterial blush precluded the procedure. Haematology input at the time of bleed warranted an apixaban-specific assay, revealing subtherapeutic levels and not requiring reversal despite the haemorrhage.
Outcome and follow-up
His mesenteric haemorrhage, however, resulted in refractory anaemia and severe multiorgan failure despite escalation to intensive care management with multiple transfusions. In view of irreversible end-organ dysfunction and ongoing clinical deterioration, his family opted for a transition into palliative care.
Discussion
Vascular events are the most feared complications of EDS. While first recognised in the 1960s by Mories on postmortem examination,4 the physician faces challenges to suspect vascular phenomenon in ‘non-vascular’ EDS given the implications on patients, their families and genetic counselling. While there is vast genetic and clinical heterogeneity of EDS, a careful assessment of bleeding tendencies is warranted in every case. In our patient, his history of spontaneous haemothorax was a hallmark feature of vascular predisposition.
There are rare reports of vascular complications in classical EDS in the literature. A systematic review of 112 papers reporting on 467 patients with non-vascular EDS showed that 77 (17%) individuals developed vascular complications.5 Overall, 3% of the individuals in the systematic review had more than one complication. Compared with vascular EDS, the classical phenotype was surprisingly associated with less frequent but more severe and life-threatening vascular complications. The most frequent vascular complication in non-vascular EDS was spontaneous or traumatic haematomas (53%). It occurred in up to 3% of patients with classical EDS-COL5A1 in the review, with most haematomas being subcutaneous but also included epidural, spinal, scalp and stomach wall. The second most common vascular complication was intracranial haemorrhage (18%), followed by dissections (16%) and aneurysms (5%). Arterial dissections are most common in medium- or large-sized arteries with a predilection for the iliac, femoral, mesenteric, brachial and coronary arteries.
Case reports of aortic dissection are relatively rare and included a left femoral and aortic aneurysm dissection in a 42-year-old man and a dissection of the infrarenal aorta and left iliac artery in a 39-year-old individual. These cases all had COL5A1 mutations.6 There are no documented cases of aortic dissection with COL5A2 mutation, which our patient had.
Genetic and acquired risk factors such as hypertension, smoking and family history of dissections independently increase the risk of genetically challenged vessel wall structures in adults with EDS. Our patient lacked any prior hypertension or family history of vascular events, including haemorrhage, aneurysms or dissections. Despite the absence of acquired risk factors, his significant smoking history would have adversely affected his vascular risk7 and fragility.
On another note, visceroptosis occurs from the displacement of abdominal viscera, manifesting in hypermobility-type EDS. It presents with varied symptoms from chronic abdominal pain, dyspepsia, bloating, dysmotility and constipation to instances of bowel obstruction with vomiting.8 Such rare manifestations of EDS should be considered particularly with chronic and progressive gastrointestinal symptoms, which were absent in our patient.
Recent evidence suggests an elevated D-dimer in acute heart failure was associated with increased stroke incidence9 and may serve as a useful biomarker in predicting stroke risk, guiding decision-making on anticoagulation. However, the evidence for the management of these vascular complications is limited and currently based on the guidelines for Marfan and Loeys-Dietz syndromes, with no EDS-subtype-specific recommendations. Celiprolol, a β-blocker, has been shown to delay vascular complications in vascular EDS.10 However, this has not been demonstrated in the classical EDS subgroups. Additionally, there are anecdotal reports of the use of desmopressin to treat and prevent bleeding in vascular EDS, but this has not been extensively studied.11 12 Hence, the management of EDS should be tailored to patient education on risk avoidance and early recognition of vascular events, allowing for early and immediate medical attention.
Learning points
Patients with Ehlers-Danlos syndrome (EDS) can have clinically heterogeneous manifestations, with red flags of vascular phenomenon of haemothorax and recurrent haematomas, predicting future risk of a catastrophic vascular event.
Traditional risk factors of smoking, hypertension and family history increase the cumulative risk of vascular phenomenon in patients with EDS.
Patient and family education on early recognition of vascular and haemorrhagic complications is warranted for prompt medical management, with counselling for the avoidance of high-risk activities and contact sports.
Ethics statements
Patient consent for publication
Footnotes
Contributors The following authors were responsible for drafting the text, sourcing and editing clinical images and investigation results, drawing original diagrams and algorithms and performing critical revisions for important intellectual content: all. The following authors gave the final approval of the manuscript: CWCC, DC and KV. CWCC is responsible for the overall content as the guarantor.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.