Article Text

Abstract
Objective Etrasimod is an oral, once-daily, selective sphingosine 1-phosphate (S1P)1,4,5 receptor modulator for the treatment of moderately to severely active ulcerative colitis (UC). S1P1 receptor expression on cardiac cells is involved in cardiac conduction. We report cardiovascular treatment-emergent adverse events (TEAEs) associated with S1P receptor modulators and other cardiovascular events in the etrasimod UC clinical programme.
Methods Patients were analysed in the Placebo-controlled UC cohort and All UC cohort. Incidence rates (IRs, per 100 patient-years) of cardiovascular-related TEAEs associated with S1P receptor modulators, including bradycardia/atrioventricular (AV) block and hypertension, and other cardiovascular events, including coronary artery disease (CAD) and cerebrovascular disease (CVD), were analysed.
Results In patients receiving etrasimod, cardiovascular-related TEAEs were infrequent (≤2.6% per AE). In the Placebo-controlled UC cohort, IRs (95% CIs) for cardiovascular-related TEAEs were higher for patients receiving etrasimod (n=577) vs placebo (n=314), respectively, for bradycardia/sinus bradycardia, 3.85 (1.58 to 6.13) vs 0 and AV block, 1.40 (0.03 to 2.76) vs 0; and numerically higher for hypertension, 5.31 (2.62 to 7.99) vs 3.40 (0.07 to 6.72). Most bradycardia/AV block events were reported on day 1. All bradycardia and hypertension TEAEs were non-serious. One serious second-degree AV block type 1 TEAE occurred in the etrasimod group; no events of second-degree AV block type 2 or higher were reported. One event each of CAD and CVD occurred in two patients receiving etrasimod.
Conclusions In the etrasimod UC clinical programme, IRs of cardiovascular-related TEAEs and other cardiovascular events were low. Most cardiovascular-related TEAEs were non-serious.
Trial registration numbers NCT02447302; NCT03945188; NCT03996369; NCT02536404; NCT03950232; NCT04176588.
- CARDIOVASCULAR COMPLICATIONS
- ADVERSE DRUG REACTIONS
- ULCERATIVE COLITIS
Data availability statement
Data are available on reasonable request. On request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual deidentified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Patients with moderately to severely active ulcerative colitis (UC) demonstrated significant improvements in clinical remission following induction and maintenance therapy with etrasimod, an oral, once-daily, selective sphingosine 1-phosphate (S1P)1,4,5 receptor modulator.
WHAT THIS STUDY ADDS
Among patients receiving etrasimod, cardiovascular treatment-emergent adverse events, including cardiac conduction-related adverse events (bradycardia/atrioventricular block) and hypertension, were infrequent, and most were non-serious.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Patients with UC have a greater risk of cardiovascular events compared with the general population, and some advanced treatments for UC are associated with an increased risk; therefore, understanding the cardiovascular adverse event risk associated with new therapies for UC is important.
Introduction
Ulcerative colitis (UC) is a chronic, immune-mediated, inflammatory disease characterised by relapsing mucosal inflammation.1 Etrasimod is an oral, once-daily (QD), selective sphingosine 1-phosphate (S1P)1,4,5 receptor modulator for the treatment of moderately to severely active UC.2 S1P mediates signalling via five isoforms of the S1P receptor (S1P1–5)3 4 and is a key regulator of lymphocyte trafficking.5 Expression of S1P1 on cardiac cells appears to play a prominent role in cardiac conduction and heart rate regulation.4 Impairment of endothelial S1P–S1P1 signalling in the resistance arteries has been associated with hypertension after chronic dosing of S1P receptor modulators, such as fingolimod.6
Patients with UC have an increased risk of cardiovascular diseases, including coronary artery diseases (CAD), such as myocardial infarction and cerebrovascular disease (CVD).7–10 In addition, therapies for UC and other inflammatory diseases have been associated with cardiovascular adverse events (AEs) including atrioventricular (AV) block and pericarditis,11 12 severe cardiovascular events13 and major adverse cardiovascular events (MACE).14 Therefore, it is important to assess cardiovascular AEs in advanced UC therapies, such as etrasimod. This integrated safety analysis aims to further contextualise the safety profile of etrasimod by describing cardiovascular events in patients with moderately to severely active UC enrolled in the etrasimod UC clinical programme.
Methods
Trial designs
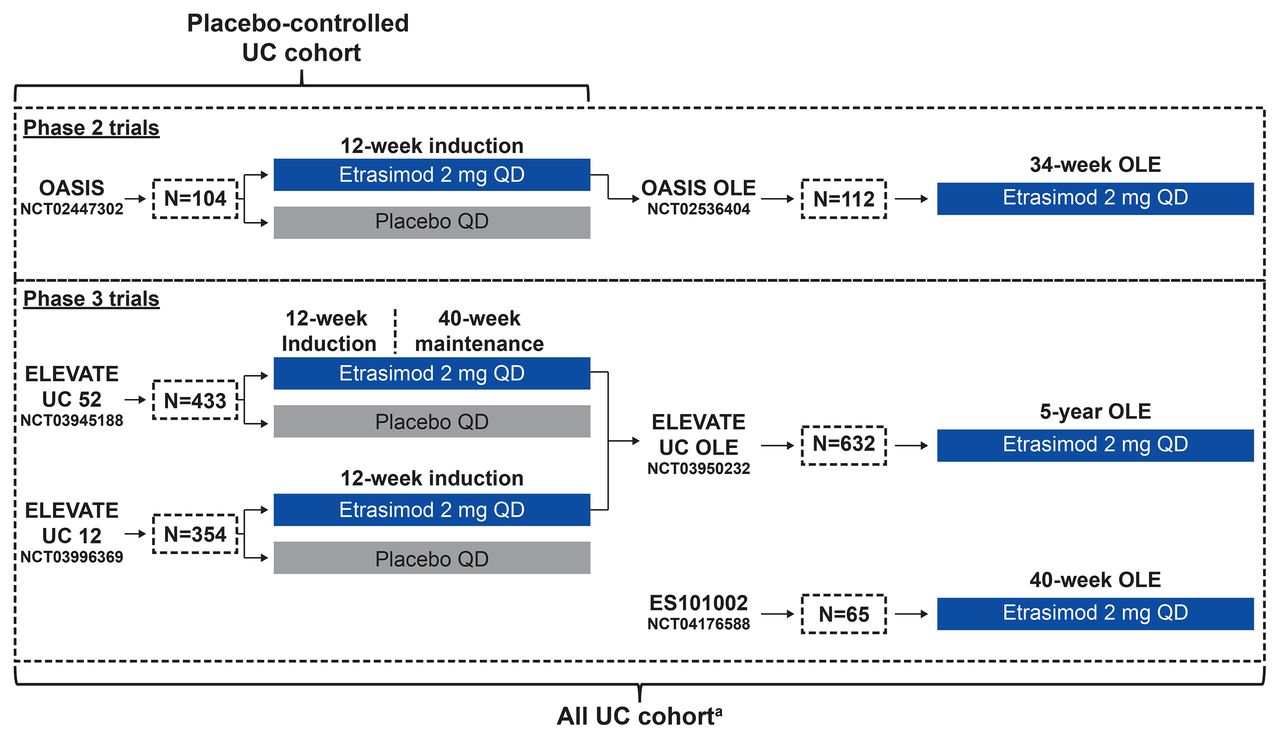
Data were pooled from patients with moderately to severely active UC in the etrasimod UC clinical programme, which consisted of a 12-week phase 2 trial (OASIS; NCT02447302), 34–40 week phase 2 open-label extension (OLE) study (OASIS OLE; NCT02536404), 52-week phase 3 induction and maintenance trial (ELEVATE UC 52; NCT03945188), 12-week phase 3 induction trial (ELEVATE UC 12; NCT03996369), ongoing 5-year phase 3 OLE trial (ELEVATE UC OLE; NCT03950232 (data cut-off: 31 January 2022)), and the open-label phase of an ongoing phase 3 trial (ES101002; NCT04176588 (data cut-off: 31 January 2022)) with a treatment period of 12–40 weeks. Study designs are shown in figure 1 with details described elsewhere.2 15 16

Study designs of trials included with pooling strategies for establishing the post hoc cohorts. aThe All UC cohort comprised only patients receiving etrasimod 2 mg once daily. N, number of patients enrolled; OLE, open-label extension; UC, ulcerative colitis.
Patients with a history of certain cardiac-related conditions/disorders were excluded from the study; full cardiac-related inclusion and exclusion criteria are listed in online supplemental table S1.
Supplemental material
Patient and public involvement
As this was a post hoc analysis, patient and public involvement was not applicable.
Analysis cohorts
Data were analysed in two cohorts, namely the Placebo-controlled UC cohort and the All UC cohort (figure 1). The Placebo-controlled UC cohort included patients treated with etrasimod 2 mg once daily or placebo in OASIS, ELEVATE UC 52 and ELEVATE UC 12. The All UC cohort comprised all patients treated with etrasimod 2 mg once daily in OASIS, OASIS OLE, ELEVATE UC 52 and ELEVATE UC 12, as well as the ongoing ELEVATE UC OLE and ES101002 (open-label phase) trials (data cut-off for both ongoing trials: 31 January 2022).
Assessments
Relevant medical history, concomitant medications and total exposure to study treatment were assessed by treatment group in each cohort. Cardiac-related screen failures were also reported for the ELEVATE UC clinical programme. The proportions of patients and exposure-adjusted incidence rates (IRs) for cardiovascular-related treatment-emergent AEs (TEAEs) of bradycardia and sinus bradycardia, AV block (including first-degree and second-degree AV block) and hypertension by preferred term (PT) were reported (Medical Dictionary for Regulatory Activities V.24.1 coding dictionary applied). Search terms for hypertension are reported in the online supplemental methods. In this study, only hypertension, blood pressure (BP) increased, hypertensive crisis, BP diastolic increased and essential hypertension events were reported.
Bradycardia, AV block and hypertension TEAEs were further medically reviewed and assessed as AEs of special interest (AESI). Cardiovascular-related TEAEs with the PT of bradycardia were considered an AESI if heart rate was <40 bpm, regardless of symptoms, <50 bpm but ≥40 bpm with symptoms or led to patient withdrawal. PTs associated with AV conduction delay were defined as an AESI if PR interval was >230 ms in first-degree AV block or AV block was second-degree (Mobitz type 1) or higher. Cardiovascular-related TEAEs (by PT) of hypertension were considered an AESI if systolic BP (SBP) was ≥160, diastolic BP (DBP) was ≥100 mm Hg or medication was added for BP control.
Cardiac conduction was measured using a 12-lead ECG prior to the first dose and at 4 hours postdose. Vital signs, including BP, heart rate, respiratory rate and temperature, were evaluated at baseline; BP and heart rate were continuously monitored hourly postdose, in the sitting position, for a total of 4 hours. Vital signs were collected at each study visit. To confirm abnormal readings, vital signs were measured up to three times during a visit. BP was determined at each visit and at 2 and 4 weeks after termination of treatment.
The proportions and IRs of other cardiovascular AEs, such as CAD and CVD, were also reported. Search terms for CAD/CVD are reported in online supplemental methods.
Statistical analysis
IRs were calculated as the number of patients with an AE divided by the total patient-years (PY) at risk for the AE, per 100 PY. PYs were defined as the sum of time to an event for patients with the particular event and censored time at-risk period for patients without such an event. The risk period was defined as the time from the first dose of study treatment to the last dose or last study visit, whichever was later. The safety analysis set was defined as all randomised patients who received ≥1 dose of study treatment in the Placebo-controlled UC cohort and the All UC cohort. For cardiovascular-related TEAEs and other cardiovascular AEs, IRs and 95% CIs were calculated using normal approximation to Poisson model, adjusted per 100 PY. If there were no events for an AE, IR was 0 and 95% CI was not calculated.
To explore potential baseline risk factors for bradycardia and sinus bradycardia, AV block and hypertension among patients receiving etrasimod 2 mg once daily in the All UC cohort, a univariate (simple) Cox analysis was used for each baseline risk factor as the only covariate. Baseline risk factors with comparison p<0.1 from simple Cox analyses were further selected in a multivariate Cox analysis model using backward selection with a model stay criterion of p<0.05. HRs and 95% CIs were calculated for comparisons of the retained baseline risk factors.
Results
Patients
Disposition, extent of exposure and screen failures
In the Placebo-controlled UC cohort, 577 patients received etrasimod 2 mg once daily and 314 received placebo. In the All UC cohort, which included open-label data, 942 patients received etrasimod 2 mg once daily. In the Placebo-controlled UC cohort, total PY of exposure to etrasimod and placebo were 276.7 (median exposure of 13.7 (range: 0.1–58.7)) and 115.1 (median exposure of 13.0 (range: 0.1–58.0) weeks), respectively. In the All UC cohort, total PY of exposure to etrasimod were 757.9 (median exposure of 37.9 (range: 0.1–132.9)) weeks.
In the ELEVATE UC pivotal trials, a total of 640 (44.8%) patients (ELEVATE UC 52, n=388; ELEVATE UC 12, n=252) failed screening. Of these, 23 (3.6%) did not meet inclusion criteria and cardiac exclusion criteria were met by a further 15 (3.9%) in ELEVATE UC 52 and 8 (3.2%) in ELEVATE UC 12 (online supplemental table S2). Screen failure data for the phase 2 trial were not included in the database.
Baseline demographics, relevant medical history and concurrent medications at baseline
Baseline demographics and cardiovascular clinical characteristics were generally similar across cohorts and treatment groups (table 1). The baseline prevalence of cardiovascular-related medical history was similar between patients treated with etrasimod and placebo, respectively, in the Placebo-controlled UC cohort (hypertension: 14.2% vs 14.3%; first-degree and second-degree AV block: 0% vs 0.3%, for both; myocardial infarction: 0.3% vs 0.3%; arrhythmia: 0.2% vs 0%). The proportions of patients with hypercholesterolaemia (4.0% vs 3.2%) and type 2 diabetes mellitus (1.6% vs 1.6%) were similar in the etrasimod and placebo treatment groups, respectively. Use of cardiovascular concomitant medications was similar across treatment groups and cohorts. For patients receiving etrasimod and placebo, respectively, in the Placebo-controlled UC cohort, the most frequently used concomitant cardiovascular medications at baseline were renin–angiotensin system agents (10.1% vs 8.0%), beta-blocking agents (5.4% vs 3.8%) and calcium channel blockers (3.8% vs 3.5%; table 1).
Baseline demographics, clinical characteristics and extent of exposure to treatment in the etrasimod UC clinical programme (Placebo-controlled UC and All UC cohorts)
S1P receptor modulator class-related cardiovascular-related TEAEs
Cardiac conduction related
Bradycardia
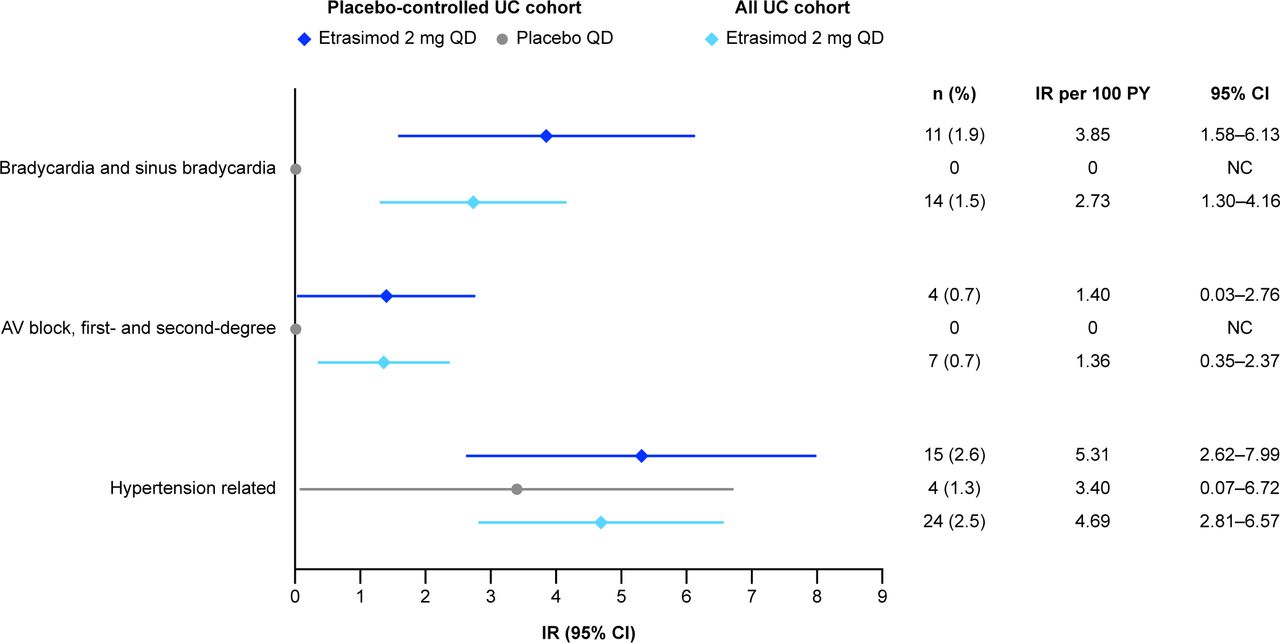
Overall, in the Placebo-controlled UC cohort, 11 (1.9%) patients receiving etrasimod and no patients receiving placebo reported TEAEs of bradycardia or sinus bradycardia. Of these cases, all were resolved, with four discontinuing treatment. In the Placebo-controlled UC cohort, the overall IR per 100 PY for bradycardia and sinus bradycardia TEAEs was higher in the etrasimod group (IR 3.85 (95% CI 1.58 to 6.13)) vs the placebo group (IR 0; figure 2). Five (0.9%) patients were further assessed for the AESI bradycardia. In the All UC cohort, 14 (1.5%) patients receiving etrasimod reported cardiovascular-related TEAEs bradycardia and sinus bradycardia (IR 2.73 (95% CI 1.30 to 4.16); figure 2); all cases were resolved, with four discontinuing treatment (same patients as in the Placebo-controlled UC cohort). No new AESIs of bradycardia were reported in the All UC cohort that were not previously reported in the Placebo-controlled cohort. The mean change from baseline in heart rate over time for all patients in the Placebo-controlled cohort is presented in online supplemental figure S1.

{kind=link}
{kind=link}
IRs per 100 PY of cardiovascular-related TEAEs in the etrasimod UC clinical programme (Placebo-controlled UC and All UC cohortsa). If there were no patients with an AE (n=0) and IR per 100 PY=0, 95% CI was not calculated and is displayed as ‘0’. aMedDRA V.24.1 coding dictionary applied. AE, adverse event; AV, atrioventricular; IR, exposure-adjusted incidence rate; MedDRA, Medical Dictionary for Regulatory Activities; n, number of patients with events within the risk period; NC, not calculated; PY, patient-years; QD, once daily; TEAE, treatment-emergent adverse event; UC, ulcerative colitis.
Baseline characteristics in patients with bradycardia and sinus bradycardia are shown in table 2. No baseline characteristics were associated with bradycardia in the Placebo-controlled and All UC cohorts.
Baseline characteristics, concomitant medications and medical history in patients with cardiovascular-related TEAEs in the etrasimod UC clinical programme (Placebo-controlled UC and All UC cohorts)
Across the etrasimod UC clinical programme, all events of bradycardia were reported on day 1 of treatment with etrasimod, aside from two events that were reported on day 2 of treatment. All bradycardia events were considered mild or moderate in severity; none were serious or associated with clinically significant haemodynamic changes. Most events of bradycardia were asymptomatic and resolved without treatment. One patient with bradycardia had a heart rate of 36 bpm and required treatment with a single intravenous dose of atropine 0.5 mg. This patient was asymptomatic throughout and remained haemodynamically stable.
Two patients had symptomatic bradycardia events. One patient with a moderate event of bradycardia also reported a moderate AE of dizziness on day 2; no treatment was administered for these events and the events resolved on day 4. One patient with a mild event of bradycardia also had non-serious mild AEs of dizziness and palpitations on day 2; these events resolved the same day and did not require treatment. Two moderate events of bradycardia in patients receiving etrasimod led to study discontinuation, one of which occurred in a patient with a history of hypertension. In addition, two mild events of bradycardia led to study discontinuation per study protocol. Characteristics of individual patients who experienced bradycardia are shown in online supplemental table S3.
Results from the exploratory multivariate Cox regression analysis demonstrated that beta-blocker (including propranolol) use at baseline may be associated with bradycardia and sinus bradycardia in the All UC cohort (HR (95% CI) 4.77 (1.07 to 21.30).
AV block
In the Placebo-controlled UC cohort, four (0.7%) patients receiving etrasimod reported the TEAE AV block (combined first-degree and second-degree; IR 1.40 (95% CI 0.03 to 2.76)), compared with zero patients receiving placebo. Of these cases, two patients discontinued treatment and one case (male, 30–40 years old; history of Hypertension) of AV block first-degree was not resolved (49-day duration) at the time of reporting. In the All UC cohort, seven (0.7%) patients receiving etrasimod reported the TEAE AV block (combined first-degree and second-degree; IR 1.36 (95% CI 0.35 to 2.37)); three patients discontinued treatment and one case (male, 30–40 years old; history of Hypertension) of AV block first-degree was not resolved by final study follow-up (49-day duration; same patient as in the Placebo-controlled UC cohort). All applicable second-degree AV block events were Mobitz type 1. No events of AV block Mobitz type 2 or higher were reported in the etrasimod UC clinical programme. Across both cohorts, all patients with TEAE AV block (combined first-degree and second-degree) met AESI criteria for AV block. The proportions and IRs of all AV block events are shown in figure 2.
Baseline characteristics in patients with AV block events are shown in table 2. Across cohorts and treatment groups, most patients with AV block were <50 years of age.
No AV block events were reported after day 2 of treatment with etrasimod; most events were considered to be mild or moderate in severity and none were associated with haemodynamic changes. One patient receiving etrasimod in the All UC cohort had a serious AE of AV block second-degree type 1, which was moderate in severity and led to study treatment discontinuation; the event did not require treatment and resolved on day 2. All AV block events, including the serious AE, were asymptomatic and resolved without treatment. One event of first-degree AV block and two events of second-degree AV block, moderate in severity, led to study discontinuation per study protocol. Characteristics of individual patients who experienced AV block are shown in online supplemental table S4.
Results from the exploratory Cox regression analyses found no baseline risk factors that were significantly associated with AV block in the All UC cohort.
Hypertension
In the Placebo-controlled UC cohort, the proportions and IRs of hypertension-related TEAEs were numerically higher in patients receiving etrasimod (n=15 (2.6%); IR 5.31 (95% CI 2.62 to 7.99)) vs placebo (n=4 (1.3%); IR 3.40 (95% CI 0.07 to 6.72)); 14 (2.4%) patients receiving etrasimod were assessed as having an AESI of hypertension. In the All UC cohort, 24 (2.5%) patients receiving etrasimod had hypertension-related TEAEs (IR 4.69 (95% CI 2.81 to 6.57)). In the All UC cohort, five (0.5%) patients receiving etrasimod were assessed as having an AESI of hypertension that had not been previously reported in the Placebo-controlled cohort. IRs of hypertension-related TEAEs are shown in figure 2.
Baseline characteristics and medical history in patients with hypertension events are shown in table 2. Across cohorts, a greater proportion of those with hypertension were ≥50 years of age. The majority of patients with hypertension events had predose SBP>120 mm Hg (Placebo-controlled UC cohort: etrasimod 2 mg once daily, 86.7%; placebo, 100.0%; All UC cohort: etrasimod 2 mg once daily, 91.7%) and DBP>80 mm Hg (Placebo-controlled UC cohort: etrasimod 2 mg once daily, 80.0%; placebo, 75.0%; All UC cohort: etrasimod 2 mg once daily, 75.0%). Half of patients with hypertension, across cohorts and treatment groups, had a history of hypertension (table 2).
Throughout the etrasimod UC clinical programme, the majority of hypertension events were mild or moderate in severity. One patient receiving etrasimod who had a history of hypertension and was receiving amlodipine at baseline, reported a TEAE of hypertension, which was considered to be severe by the investigator; this event started on study day 169 and was determined to be unrelated to study treatment by the investigator. In addition, 16 of 20 (80.0%) patients receiving etrasimod who experienced hypertension events initiated treatment with anti-hypertensive medications; nine (56.3%) of these patients did not have a medical history of hypertension. No patients discontinued the study due to hypertension (online supplemental table S5).
A moderate TEAE of hypertensive crisis was reported in one patient in ELEVATE UC 52 by the investigator on day 218. The patient had independently self-administered one dose of captopril, but no recorded BP was available, and the patient was not able to substantiate that there was elevated BP on this date. Vital signs prior to and following this event were all normal. No changes were made to study treatment and the event was resolved on the same day without intervention from the investigator. This event was confirmed by the investigator as not being a ‘hypertensive crisis’ event based on the objective BP measurements and accepted diagnostic definitions.
Among all patients with hypertension-related TEAEs in the Placebo-controlled UC cohort and All UC cohort, small changes from baseline in SBP (−20.0 to 12.6 mm Hg) or DBP (−10.5 to 5.0 mm Hg) were observed in patients treated with etrasimod vs placebo (online supplemental table S6).
Results from the exploratory multivariate Cox regression model demonstrated that age group (≥50 vs <50 years of age) may be associated with hypertension in the All UC cohort (HR (95% CI) 3.93 (1.72, 9.00)).
Other cardiovascular AEs
The incidence of CAD and CVD was generally low across cohorts and treatment groups (table 3). In the Placebo-controlled UC cohort, one (0.2%) patient each reported CAD (PT: CAD) and CVD (PT: Carotid arteriosclerosis) in the etrasimod group (IR 0.35 (95% CI 0.00 to 1.03) for both CAD and CVD). No patients in the placebo group reported CAD or CVD. The same patients reported CAD (PT: CAD) and CVD (PT: Carotid arteriosclerosis) in the etrasimod group in the All UC cohort (IR 0.19 (95% CI 0.00 to 0.57 for both CAD and CVD); table 3). Two (0.2%) patients experienced a TEAE of transient ischaemic attack (one event each) in the All UC cohort (IR 0.26 (95% CI 0.00 to 0.61)). No additional cases were reported among patients treated with etrasimod in the All UC cohort.
Proportions and IRs per 100 PY of CAD and CVD in the etrasimod UC clinical programme
Across cohorts, both patients with CAD/CVD TEAEs were ≥50 years of age with SBP>120 mm Hg and DBP>80 mm Hg and had a history of hypertension at baseline (online supplemental table S7). One patient in the All UC cohort reported CVD, which was considered to be moderate in severity and did not lead to study discontinuation. One patient in the All UC cohort, with medical history of dyslipidaemia and hypertension, reported CAD, which was a serious AE; this event was assessed as not related to the treatment by the investigator and did not lead to study discontinuation.
Discussion
Understanding cardiovascular events among patients with UC is important as disease states and some treatment options incur an increased risk.7 14 This integrated safety analysis evaluated the occurrence of cardiovascular-related TEAEs among patients with moderately to severely active UC in the etrasimod UC clinical programme.
Cardiovascular-related TEAEs were infrequent in patients treated with etrasimod. All reported bradycardia (1.9% and 1.5% for the Placebo-controlled and All UC cohorts) and AV block (0.7% for the Placebo-controlled and All UC cohorts) events were transient and mild or moderate in severity, and most were non-serious, reported on day 1 post-first dose and was resolved without treatment. Patients were generally haemodynamically stable throughout, and few events of bradycardia were accompanied by symptoms; no events of clinical consequence, such as syncope or fall, were associated with bradycardia events. One AV block event (second-degree Mobitz type 1) met seriousness criteria but did not require treatment intervention and was resolved on day 2. The majority of patients did not discontinue treatment due to cardiovascular-related TEAEs.
Cardiac conduction abnormalities, including bradycardia and AV block, have been observed within hours of first-dose administration with S1P receptor modulators. For several S1P receptor modulators, including ozanimod, these reported conduction events have necessitated a dose titration.4 15 17 18 Due to the transient and modest first-dose effect of etrasimod, and potentially its low affinity for S1P3 receptors (which may contribute to bradycardia) compared with non-selective S1P receptor modulators,19 no dose titration was implemented in the etrasimod UC clinical programme.2 15 The incidence of bradycardia and AV block in patients treated with etrasimod without dose titration was comparable to that observed with other S1P receptor modulators that required a dose-titration phase.2 20 21
Cardiovascular safety results from the All UC cohort demonstrate that the incidence of TEAEs of bradycardia was low (n=14 (1.5%)); mostly mild, and all but two events were reported on day 1, with no new events reported after day 2. Likewise, the incidence of TEAEs of AV first-degree and second-degree block (Mobitz type 1) was low in the All UC cohort (n=7 (0.7%)); findings are similar to those reported for ozanimod. In a post hoc analysis of the phase 3 True North and OLE studies in patients with moderately to severely active UC, bradycardia occurred more frequently with ozanimod versus placebo (ozanimod: n=6 (0.6%); placebo: n=0),22 with two events reported on day 1 during the first 6 hours of treatment.22 Of the three patients who reported bradycardia after day 1 of treatment with ozanimod, one was symptomatic and discontinued ozanimod, one required treatment intervention with meldonium and discontinued ozanimod, and one recovered without intervention.22 Results from the exploratory multivariate Cox regression analysis may suggest that the use of beta-blockers (including propranolol) at baseline is associated with bradycardia and sinus bradycardia in the All UC cohort; however, interpretation of this analysis is limited due to the small sample size (N=32) and that it does not account for baseline/predose HR, which may be lower in patients taking beta-blockers. Regardless, patients taking beta-blockers should be thoroughly assessed prior to initiating etrasimod treatment. Further controlled studies and investigations with larger exposure profiles may be needed.
Throughout the etrasimod UC clinical programme (2.5 years), few hypertension events were reported. No affected patients discontinued due to hypertension, and the risk of occurrence did not increase over time. Furthermore, in the All UC cohort, most patients with hypertension events had baseline BP>120/80 mm Hg. In the ELEVATE UC clinical programme, small increases in SBP were reported in patients receiving etrasimod versus placebo while no clinically meaningful changes in DBP were reported in either treatment group over 52 weeks of treatment.
When compared with healthy individuals, patients with active UC are at a higher risk of developing cardiovascular diseases due to chronic inflammation, which plays a pivotal role in atherogenesis.23 Historically, some therapies for the treatment of chronic inflammatory diseases have been associated with an increased risk of other cardiovascular AEs.24 25 Results from the postauthorisation safety study in risk-enriched rheumatoid arthritis patients (ORAL Surveillance) indicated a higher frequency of MACE with tofacitinib (IR 0.98 (95% CI 0.79 to 1.19)) vs tumour necrosis factor inhibitor (IR 0.73 (95% CI 0.52 to 1.01)).14 It should be noted that patients enrolled in ORAL Surveillance were ≥50 years of age, whereas the majority of patients in the etrasimod UC clinical programme were <50 years of age. However, in a post hoc analysis of the tofacitinib UC OCTAVE clinical programme, the overall IR of MACE was 0.29 (95% CI 0.13 to 0.55), similar to real-world incidence for patients with UC.26 Ustekinumab was also significantly associated with an increased risk of acute coronary syndrome and stroke among patients with psoriasis who were at high cardiovascular risk (OR 4.17 (95% CI 1.19 to 14.59)).13 Furthermore, in a pooled safety analysis of ustekinumab, the IR of serious MACE was 0.16 (95% CI 0.00 to 0.89) in patients with UC.27 Compared with Janus kinase inhibitors, S1P receptor modulators are not typically associated with MACE28; this is in line with our findings as low overall IRs of CAD and CVD were observed in patients treated with etrasimod throughout the etrasimod UC clinical programme.
There are some limitations in this analysis. Patients included in this integrated safety analysis were eligible based on specific inclusion and exclusion criteria, including cardiac criteria, and therefore, may not represent the overall UC population. Notably, the eligibility criteria for the etrasimod UC clinical programme excluded patients with certain cardiovascular conditions, which may account for the low incidence of cardiovascular-related TEAEs. Data interpretation is also limited by the small sample size of patients who experienced cardiovascular events. Findings from the exploratory multivariate Cox analyses should be interpreted with caution as these were not controlled for type 1 errors.
In conclusion, in patients with moderately to severely active UC treated with etrasimod, cardiovascular-related TEAEs associated with S1P receptor modulators, including cardiac conduction-related AEs (bradycardia/AV block) and hypertension, were infrequent. Low rates of other cardiovascular AEs, namely CAD and CVD, were observed in the patients who received etrasimod in the UC clinical development programme. Most cardiovascular-related TEAEs were considered non-serious. Across the phase 2 and phase 3 trials, the cardiovascular safety profile of etrasimod during treatment initiation was similar to that of other S1P receptor modulators that require a dose-titration phase. Future results from the ongoing ELEVATE UC OLE trial will provide valuable insights into the cardiovascular safety profile of etrasimod, particularly longer-term safety data, in patients with moderately to severely active UC.
Data availability statement
Data are available on reasonable request. On request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual deidentified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and all studies were registered with ClinicalTrials.gov and were conducted in compliance with the Declaration of Helsinki and were approved by the Institutional Review Boards at each investigational centre participating in the studies. This full list of participating sites can be found on ClinicalTrials.gov via the trial identifiers NCT02447302, NCT03945188, NCT03996369, NCT02536404, NCT03950232 and NCT04176588. All patients provided written informed consent; a parent or legal guardian provided consent for patients <18 years of age who participated in the study. Further details are included in the original publications. Participants gave informed consent to participate in the study before taking part.2 15 29
Acknowledgments
The authors would like to thank the patients, investigators and study teams involved in the etrasimod UC clinical programme. Christopher J Rabbat (an employee of Pfizer at the time of the analysis) contributed to the design of the study and the interpretation of data and contributed as an author on congress abstracts and presentations related to this analysis. This study was sponsored by Pfizer. Medical writing support, under the direction of the authors, was provided by Mark Bloom, PhD and Megan Melody, MSc, CMC Connect, a division of IPG Health Medical Communications, and was funded by Pfizer, New York, New York, USA, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med 2022;175:1298–304).
References
Footnotes
Contributors JW, ABD and AJY contributed to the concept and methodology; AJY contributed to data curation; GG and AJY contributed to the investigation; JW and ABD contributed to data validation; GG, JW, XG and ABD contributed to the formal analysis; SV, DTR, LPB, MCD, MR, PMI, MG, KL, GG, JW, IM, AM, XG, JG, ABD and AJY contributed to the writing, reviewing and editing of the manuscript. All authors approved the final version of the manuscript, including the authorship list. XG is the guarantor.
Funding This work was supported by Pfizer (Grant no: NA). The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: this study was sponsored by Pfizer. Funding for medical writing support was provided by Pfizer.
Competing interests SV has received lecture/speaker fees from AbbVie, Dr. Falk Pharma, Ferring Pharmaceuticals, Hospira, MSD, Takeda and Tillotts; has served as a consultant and advisory board member for AbbVie, AbolerIS Pharma, Alimentiv, Arena Pharmaceuticals, AstraZeneca, Avaxia, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, CVasThera, Dr. Falk Pharma, Eli Lilly, Ferring Pharmaceuticals, Galapagos, Genentech/Roche, Gilead Sciences, Hospira, Imidomics, Janssen Pharmaceuticals, Johnson and Johnson, Materia Prima, MiroBio, Morphic, MrMHealth, MSD, Mundipharma, Pfizer, Prodigest, Progenity, Prometheus Biosciences, Robarts Clinical Trials, Second Genome, Shire, Surrozen, Takeda, Theravance Biopharma, Tillotts Pharma AG and Zealand Pharma; has received grant/research funding from AbbVie, Galapagos, MSD, Pfizer and Takeda. DTR has served as a consultant and advisory board member for AbbVie, Altrubio, Aslan Pharmaceuticals, Athos Therapeutics, Bellatrix Pharmaceuticals, Bristol Myers Squibb, Celgene, Chronicles, ClostraBio, Genentech/Roche, Gilead Sciences, Iterative Health, Janssen Pharmaceuticals, Lilly, Pfizer, Prometheus Biosciences, Reistone, Seres Therapeutics, Takeda, Target RWE and Trellus Health; has received grant/research funding from GastroIntestinal Research Foundation, Helmsley Charitable Trust and Takeda; has served on the board of trustees for Crohn’s & Colitis Foundation and Cornerstones Health; owns stock in Altrubio, Datos Health, and Iterative Health. LPB has received grant/research funding from Celltrion, Fresenius Kabi and Takeda; has served as a consultant for AbbVie, Abivax, Alimentiv, Alma Bio Therapeutics, Amgen, Applied Molecular Transport, Arena Pharmaceuticals, Biogen, Bristol Myers Squibb, Celltrion, CONNECT Biopharm, Cytoki Pharma, Eli Lilly, Enthera, Ferring Pharmaceuticals, Fresenius Kabi, Galapagos, Genentech, Gilead Sciences, Gossamer Bio, GSK, HAC-Pharma, IAG Image Analysis, Index Pharmaceuticals, Inotrem, Janssen Pharmaceuticals, Medac, Mopac, Morphic, MSD, Norgine, Novartis, OM Pharma, ONO Pharma, OSE Immunotherapeutics, Pandion Therapeutics, Par'Immune, Pfizer, Prometheus Biosciences, Protagonist, Roche, Sandoz, Takeda, Theravance Biopharma, Thermo Fisher, TiGenix, Tillotts, Viatris, Vifor and Ysopia; has received lecture/speaker fees from AbbVie, Amgen, Arena Pharmaceuticals, Biogen, Celltrion, Eli Lilly, Ferring Pharmaceuticals, Galapagos, Genentech, Gilead Sciences, Janssen Pharmaceuticals, Medac, MSD, Pfizer, Sandoz, Sanofi, Takeda, Tillotts, Viatris and Vifor; owns stock in CTMA. MCD has served as a consultant for AbbVie, Abivax, Arena Pharmaceuticals, AstraZeneca, Bristol Myers Squibb, Celgene, Eli Lilly, Galapagos, Genentech, Gilead Sciences, Janssen Pharmaceuticals, Pfizer, Prometheus Laboratories, Prometheus Biosciences, Takeda, and UCB; has received grant/research funding from Janssen; has shareholder/royalties in Trellus Health; has directorship/ownership interest in Trellus Health. MR has received unrestricted educational grants from AbbVie, Bristol Myers Squibb, Celgene, Genentech, Gilead Sciences, Janssen Pharmaceuticals, Pfizer, Takeda and UCB; has served as a consultant and advisory board member for AbbVie, Alfasigma S.p.A., Allergan, Amgen, Bristol Myers Squibb, Celgene, Genentech, Gilead Sciences, Lilly, Janssen Pharmaceuticals, Miraca Labs, Pfizer, Prometheus Biosciences, Salix, Seres, Takeda, TARGET Pharma Solutions and UCB; continuing medical education companies: CME Outfitters, Cornerstones, GI Health Foundation (GiHF), Imedex, MJH life sciences, and Remedy; has royalties in Wolters Kluwer Health. PMI has received grant support from Celltrion, Galapagos, MSD, and Takeda; has received lecture fees from AbbVie, Bristol Myers Squibb, Celgene, Celltrion, Dr. Falk Pharma, Ferring Pharmaceuticals, Galapagos, Gilead Sciences, Janssen Pharmaceuticals, MSD, Pfizer, Sandoz, Sapphire Medical, Shire, Takeda, Tillotts and Warner Chilcott; has served as an advisory board member for AbbVie, Arena Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Celltrion, Genentech, Gilead Sciences, Hospira, Janssen Pharmaceuticals, Lilly, MSD, Pfizer, Pharmacosmos, Prometheus Biosciences, Roche, Samsung Bioepis, Sandoz, Takeda, Topivert, VH2, Vifor Pharma and Warner Chilcott. MG, KL, GG, JW, IM, AM, XG, JG and ABD are employees and shareholders of Pfizer. AJY has served as a consultant for AbbVie, Arena Pharmaceuticals, Bristol Myers Squibb, Pfizer and Takeda; has received lecture/speaker fees from Bristol Myers Squibb.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.