Article Text

Abstract
Incretins such as glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are intestinal postprandial hormones that stimulate insulin release from the pancreas as long as circulating glucose concentrations are raised. In addition to their effect on insulin secretion and consequent glucose lowering, GIP and GLP-1, especially the latter, have a number of physiological effects such as inhibition of glucagon release, gastric emptying and food intake, as well as a tropic action on pancreatic B-cell mass. There is currently a pandemic of obesity and diabetes, and existing treatments are largely inadequate both in regard to efficacy as well as their ability to tackle important factors in the pathogenesis of type 2 diabetes (T2D). There is increasing evidence that current treatments do not address the issue of progressive B-cell failure in T2D. Since obesity is the engine that is driving the epidemic of diabetes, it is disappointing that most treatments that succeed in lowering plasma glucose are also associated with weight gain. It is now well established that intensively treated T2D has a better outcome than standard treatment. Consequently, achieving better control of diabetes with lower HbA1c is the goal of optimal treatment. Despite the use of usual therapeutic agents in T2D, often in high doses and as combinations, such as metformin, sulphonylurea, α-glycosidase inhibitors, thiazolidinediones and a number of animal and human insulin preparations, optimal control of glycaemia is not achieved. The use of incretins as therapeutic agents offers a new approach to the treatment of T2D.
Statistics from Altmetric.com
There is evidence that incretin metabolism is abnormal in type 2 diabetes mellitus (T2D). The incretin effect, which can be described as the difference in insulin secretion to an intestinally administered glucose compared to that observed when the same amount of glucose is given intravenously, is markedly impaired in T2D. Secretion of GIP and GLP-1 in T2D in response to ingestion of nutrients is inappropriately low. GIP has proved to be disappointing in therapeutic use since its intravenous administration in T2D is not associated with improvement in insulin secretion and glucose tolerance, suggesting that resistance to GIP is a feature of this disease. GLP-1 on the other hand when administered intravenously in T2D is able to increase insulin secretion and improve glucose homoeostasis. GIP and GLP-1 are peptide hormones with very short half-lives and are degraded mainly by the enzyme dipeptidyl peptidase IV (DPPIV). Analogues of GIP and GLP-1 that are resistant to the action of DPPIV have been developed and clinical trials have shown their effectiveness. Another novel agent, naturally resistant to DPPIV, that is given by subcutaneous injection is a synthetic peptide, exenatide, which has recently been approved for treatment of T2D in the USA. Efforts are underway to develop agents that can be given orally. They include a DPPIV inhibitor that has been licensed for the treatment of T2D in the USA; several other agents are undergoing clinical trials. However, DPPIV is an enzyme that is involved in the degradation of a number of biological compounds and there is consequently a concern about their long-term safety. Strategies to augment the biological actions of GIP and/or GLP-1 are expected to minimise weight gain, reduce hypoglycaemic episodes and prevent progressive B-cell failure by increasing B-cell mass. The optimal agent(s) that may mimic and replace the endogenous incretin effect is not fully known and awaits the outcome of ongoing clinical trials. Important issues with regard to practicalities, such as oral or parenteral route of administration, longer term safety, and efficacy may determine which of the proposed options to increase the incretin effect is preferred in clinical practice.
T2D is an increasingly common chronic disease, consuming significant health resources; it currently affects nearly 170 million people worldwide, a number estimated double by 2030.1 Rising obesity is fuelling this alarming increase in T2D, leading to clinically significant morbidity and decreased survival. Poor glycaemic control accounts for much of the increase in morbidity, with an increased risk of microvascular and macrovascular complications.2 The United Kingdom Prospective Diabetes Study (UKPDS) reported that glycaemic control deteriorates over time, even when T2D is intensively treated.3 Despite the use of improved lifestyle measures and multiple therapeutic agents, glycaemic control progressively worsens, and more than 60% of people with diabetes have poor diabetic control, as reflected by HbA1c values above 7%.4 There is therefore a need for a new approach to the treatment of T2D. An approach using “incretin” therapy is promising; the basis for this approach is described in this short review.
GLUCOSE METABOLISM IN HEALTH
T2D is primarily a derangement in glucose homoeostasis and a consideration of normal glucose metabolism is in order. Humans are periodic feeders, spending most of the day in the postprandial period, and consume approximately 50% of their daily caloric requirement as carbohydrate. The digestion and absorption of carbohydrate increases circulating glucose concentration and requires insulin for proper storage and metabolism. A rise in arterial glucose perfusing the pancreatic B-cell leads to insulin release, which in turn restores euglycaemia. In addition to this direct effect of glucose on the B-cell, intestinal factors have a major role in the regulation of pancreatic endocrine secretion since oral rather than intravenous glucose administration results in greater insulin secretion.5 The factors released from the gut in response to glucose absorption, such as glucose-dependent insulinotropic polypeptide6 (also known as gastric inhibitory polypeptide or GIP) and glucagon-like peptide-1 (or GLP-1)7 sensitise the B-cell of the pancreas and reduce the threshold for release of insulin; they form part of the entero-insular axis (fig 1). Only around 20–30% of the oral glucose-induced insulin secretion is accounted for by GIP, GLP-1 accounting for the remainder.8

INCRETIN CONCEPT
The difference in the insulin secretion between an amount of glucose given orally and the same amount given intravenously is called the “incretin effect”, and the hormones responsible are called “incretins” (⇓ figs 2 and 3). The following criteria have to be fulfilled for an agent to be called an incretin: (1) it must be released in response to oral nutrient ingestion, especially glucose; and (2) it must reach physiological concentrations in vivo to cause insulin release. GLP-1 and GIP are currently the only known incretins. A further property of these incretins that make them attractive as potential therapeutic agents in diabetes is that insulin secretion to these peptide hormones ceases when euglycaemia is achieved, thereby minimising the risk of hypoglycaemia.9 ,10


BIOLOGY OF INCRETINS (TABLE 1)
Cells of origin
GIP gene is expressed mainly in the enterochromaffin cells (K-cells) of the proximal small intestine. The proglucagon gene is expressed in a type of enterochromaffin cell, the L-cells of the intestine, where further processing of the peptide by proconvertase 1/3 leads to GLP-1 production.11 Processing of the proglucagon gene product by proconvertase 2 in the pancreatic A-cells of the islets leads to glucagon production.
Structure of incretins
GIP is produced as a 42 amino acid peptide, whereas GLP-1 is produced as an inactive 37-amino acid peptide whose C-terminal end contains glycine (fig 4). The active form is produced by post-translational cleavage of six amino acids from the N-terminal end of GLP-1(1–37). This truncated (7–37) form of GLP-1 can be amidated at the glycine end of the C-terminal and is the major circulating form. Both GLP-1(7–37) and GLP-1(7–36 amide) are equipotent insulinotropic peptides.

Incretin receptors
GIP receptors are expressed in a number of cell types including pancreatic B and A cells, stomach, adipose tissue and brain. GLP-1 receptors are found on B- and D-cells of the pancreas, parietal cells of the stomach, pylorus, adipose tissue, lungs and the brain.11
REGULATION OF INCRETIN SECRETION
Stimuli for incretin release
Ingestion of nutrients such as carbohydrate, protein and fat, leads to release of GLP-112 as well as GIP; of these, carbohydrate ingestion is the best stimulus for GLP-1 secretion. Only carbohydrate absorption mediated GLP-1 secretion leads to insulin secretion, whereas fat- and protein-mediated GLP-1 secretion do not.
Kinetics of GLP-1 secretion
Circulating GIP and GLP-1 concentrations rise within 15 minutes following ingestion of nutrients; peak concentrations of GIP and GLP-1, around 200 and 50 pmol/l respectively, are attained by 30–45 minutes, returning to basal values by 2–3 hours. Following secretion, both GIP and GLP-1 have a very short circulating half-life of 3–5 minutes due to the action of proteases such as dipeptidyl peptidase IV (DPPIV) and other endopeptidases, resulting in inactivation.13
Dipeptidyl peptidase IV
DPPIV is a ubiquitous enzyme present on all cell surfaces and body fluids, with a diverse number of substrates that includes GIP and GLP-1. The action of DPPIV on GIP and GLP-1 (7–36 amide) leads to the removal of two N-terminal amino acids, resulting in the formation of GIP 3–42 and GLP-1(9–36 amide), both the de-amidated peptides acting as antagonists at their respective receptors.
Factors affecting GLP-1 secretion
Nutrient intake, composition and absorption are major factors affecting release of GLP-1. The rate of nutrient entry into the small intestine, determined by gastric emptying, may further modulate secretion of GIP and GLP-1. A rapid gastric emptying as evidenced by post-gastrectomy dumping syndromes causes marked increase in GIP and GLP-1 secretion.14 Mechanical factors, such as osmotic phenomena or distension, may also enhance secretion of GLP-1 such as that seen following administration of the α-1-glycosidase inhibitor acarbose.15 Apart from intestinal luminal factors, systemic factors may also modulate GIP and GLP-1 secretion. Circulating free fatty acids,16 as well as pancreatic glucagon,17 both predominantly present during the fasting state, suppress the secretion of GLP-1.
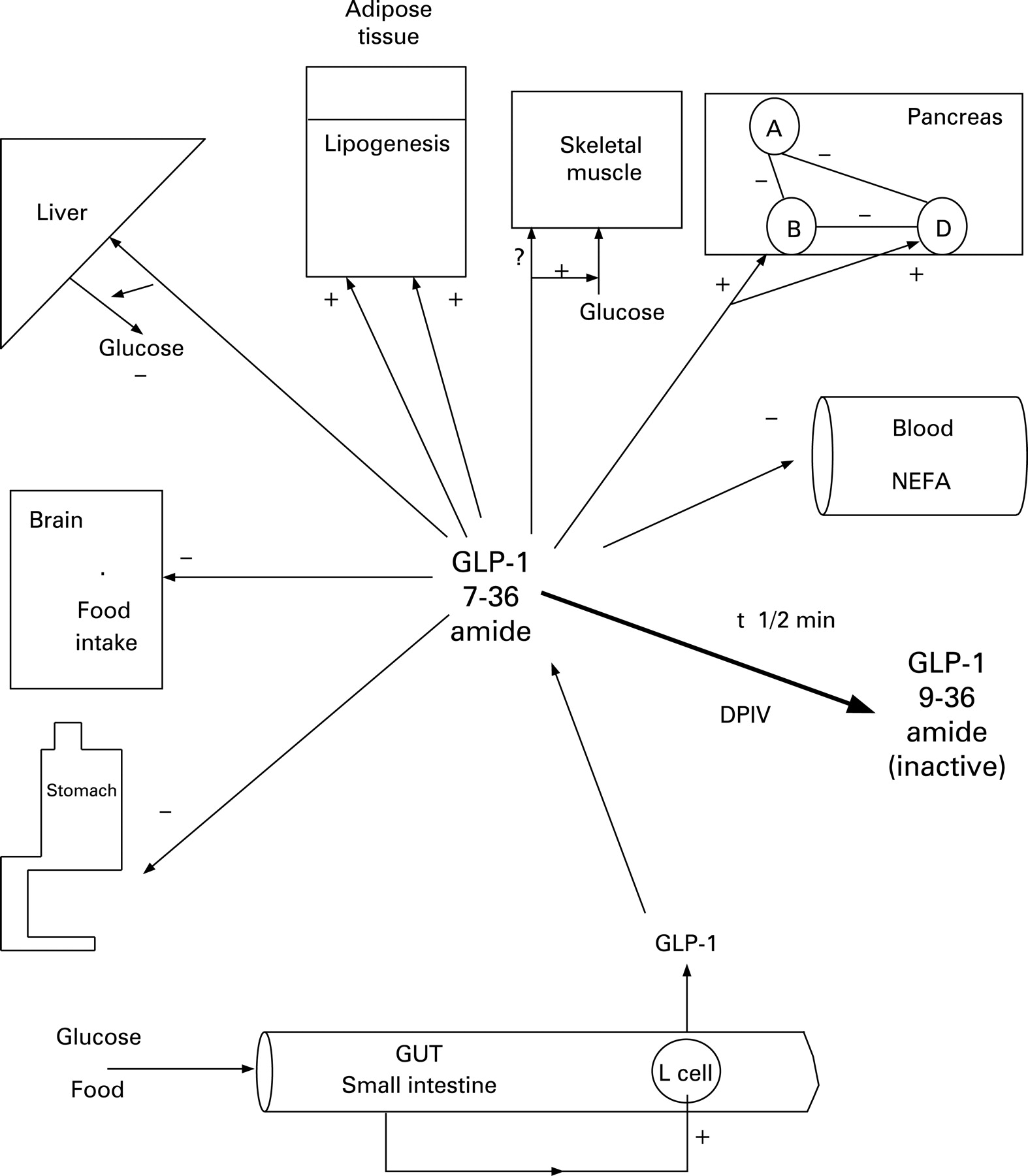
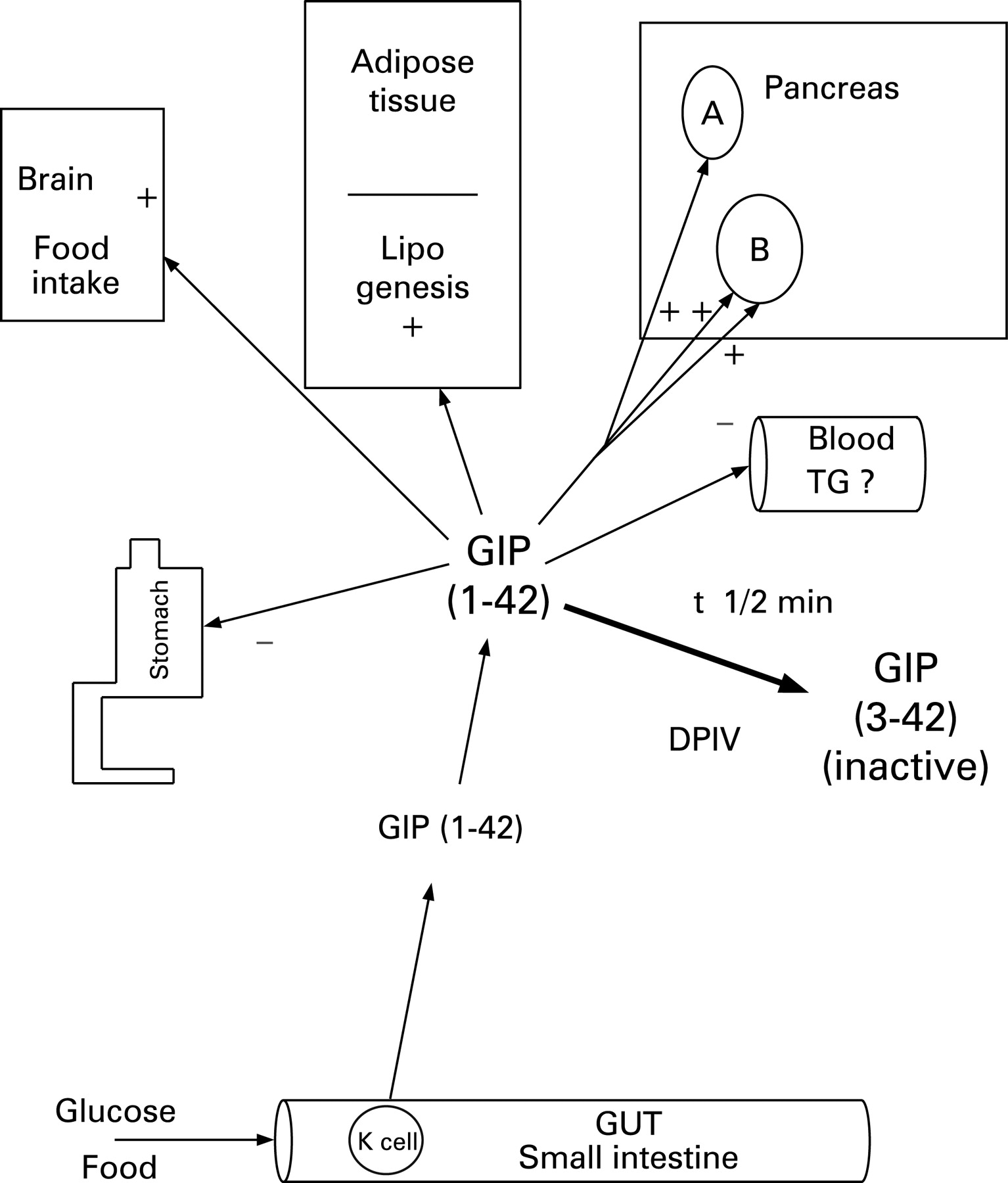
EFFECTS OF GIP AND GLP-1 (⇓FIGS 5 AND 6)
Insulin secretion
The most important property of GIP and GLP-1 in clinical terms is its ability to promote insulin secretion and maintain glucose homoeostasis without inducing hypoglycaemia.18 GLP-1 acts on the pancreatic B-cell by binding to a specific receptor that is coupled to glucose entry and metabolism, allowing the manifestation of its glucose-dependent property.19 In GIP and/or GLP-1 knockout mice, diabetes mellitus was the main consequence of a lack of incretin effect.20
Glucagon secretion
Pancreatic glucagon secretion is either unaffected or increased by GIP.21 Although there are no receptors for GLP-1 on the A-cell, intra-islet insulin released from B-cells in response to GLP-1 locally inhibits glucagon secretion. The resulting improvement in insulin/glucagon ratio may account at least partly for the reduction in insulin resistance, in both the liver and peripheral tissues, such as the skeletal muscle.22
Somatostatin secretion
Direct receptor-mediated stimulation of the D-cell by GLP-1 has been observed,23 indicating a complex interplay between GLP-1 and all the major endocrine cells of the islets that may be important in the fine-tuning of homoeostatic control of glucose metabolism.
B-cells mass
GLP-1 has a tropic effect on B-cells, both in terms of enhancing the magnitude of insulin secretion as well as increasing their number. GLP-1 increases B-cell mass by stimulating proliferation and induction of islet neogenesis as well as by inhibiting apoptosis.24 GLP-1 also promotes cell differentiation, from exocrine ductal cells or immature islet progenitors, towards a more differentiated B-cell phenotype.25 GIP has also been shown to increase B-cell number and mass.26
Stomach
Receptors for GIP and GLP-1 are present in the stomach. Stimulation of GLP-1 receptors in the pyloric sphincter delays gastric emptying27 and minimises postprandial glycaemia. By delaying gastric emptying, it also influences the distension of the stomach and peripheral satiety signals. Like GIP, the effect of GLP-1 on gastric acid secretion at physiological concentrations in vivo is disputed.
Brain
Although GLP-1 synthesis occurs in the brain, peripheral GLP-1 may bind to regions of the brain where the blood–brain barrier is deficient; these include the brain centres controlling food intake and energy expenditure.28 GLP-1 may mediate satiety effects at least in part through this mechanism. GIP receptor expression has also been found in the brain; GIP administration in healthy subjects appears to increase food intake and decrease energy expenditure, in contrast to GLP-1.29 ,30

PATHOPHYSIOLOGY OF T2D
Traditionally, insulin resistance and B-cell failure are two core defects that have been suggested to explain the pathophysiology of T2D. Hyperglycaemia, representing a balance between circulating glucose appearance and consumption, is the cardinal feature of T2D. In health, insulin and glucagon are the key regulators of glucose appearance and consumption during the postprandial and the fasting periods respectively,31 and their abnormality is believed to explain the disordered glucose metabolism in T2D. In patients with T2D, the rate of hepatic glucose production is inappropriately increased, despite raised insulin concentrations, and indicates hepatic insulin resistance. The ability of endogenous insulin to increase plasma glucose uptake by peripheral tissues is markedly reduced, and this reduction indicates peripheral insulin resistance.32 Oral antihyperglycaemic agents that reduce basal hepatic glucose production and enhance glucose uptake in skeletal muscle (e.g. thiazolidinediones, biguanides) by means of increasing sensitivity to insulin, form a crucial part of current diabetes management.33 Table 2 presents a summary of the effects of drugs currently available for use in T2D on the pathophysiological defects of the disease.
PATHOPHYSIOLOGICAL FACTORS IN T2D NOT ADDRESSED BY CURRENT TREATMENT
Decreased incretin effect (table 3)
During the evolution of T2D, insulin resistance stabilises early, becoming constant; initially pancreatic B-cells can maintain normal plasma glucose concentrations by compensatory hyperinsulinaemia. Overt T2D occurs only when B-cells numbers fall or become dysfunctional and can no longer maintain the hyperinsulinaemia required to maintain normoglycaemia.32 Early in the natural history of T2D, compensatory hyperinsulinaemia is successful in overcoming the insulin resistance and maintaining euglycaemia. Failure of incretin effect leads to relative insulinopenia and hyperglycaemia. GLP-1 secretion is reduced in non-diabetic obese subjects, suggesting that incretin secretion may be altered early in the natural history of diabesity.34 A number of investigators have documented a reduced incretin effect in T2D.35 ,36 This finding implies that signals from gut-derived factors are attenuated because of decreased concentrations of incretin hormones or resistance to their effects. Studies on GIP secretion in T2D report decreased, normal and increased responses.11 Earlier studies on secretion of GLP-1 in T2D suggested that circulating concentrations are raised, but recent studies employing better control of metabolic factors and more specific analytical assays indicate a near absence.37

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Weight gain
Most T2D patients are overweight and most current treatments, apart from the biguanides, do not produce weight loss. In fact the weight gain associated with treatments such as the sulphonylureas and insulin may reduce the overall effectiveness of such approaches and worsen outcome in the long run.38 – 40
Decreased B-cell mass
The UKPDS data suggest that newly diagnosed T2D have only 50% B-cell function, declining further thereafter.41 An early marker of B-cell dysfunction, the loss of first-phase insulin secretion is present before diabetes is diagnosed, when a patient has impaired glucose tolerance. The loss impairs early prandial suppression of hepatic glucose production and exaggerates already raised postprandial glucose levels. Amelioration of the decline in B-cell function must be addressed to alter the progressive nature of the disease. The progressive decline in B-cell function has been attributed to hyperglycaemia, causing glucotoxicity, and increased circulating non-esterified fatty acids (NEFA), causing lipotoxicity.42 ,43
Dysregulation of gastric emptying
Gastric emptying may be more rapid in T2D and may be a key determinant of postprandial glucose excursions.44 ,45 A slowing in gastric emptying during hyperglycaemia occurs to improve the match between rise in glucose due to nutrient absorption and glucose disappearance from the circulation, despite which, the gastric half-emptying time is significantly shorter in patients with T2D. Two new therapies, amylin (and its analogue pramlintide) and incretin mimetics, can modify the gastric-emptying rate.
Hyperglucagonaemia
Increased circulating glucagon in T2D is not suppressed after a carbohydrate-rich meal and occurs early in prediabetes.46 – 48 The resultant hyperglucagonaemia contributes both to an inability to suppress hepatic glucose production postprandially and to excessive plasma glucose excursions. Current therapies do not influence the hyperglucagonaemia and an approach that targets glucagon suppression would offer a further mechanism to improve glycaemic control.
POTENTIAL NEW TREATMENTS FOR T2D
GIP administration appears to be ineffective in the treatment of T2D49; this is likely to be due to B-cell resistance to this peptide. Therefore current approaches to incretin therapy are based on enhancing the biological effect at the GLP-1 receptor50 ,51 (table 4).
At least two different approaches to augmentation of the incretin effect can be used in therapy. One would be to administer incretin(s) exogenously; there are a number of potential agents with differing properties and issues. An alternate strategy would be to enhance endogenous incretins.
AUGMENTATION OF ENDOGENOUS GLP-1
Inhibition of intestinal α-glycosidase has been shown to augment secretion of both GIP and GLP-1.14 Although α-glycosidase inhibition by drugs such as acarbose and meglitol is used in the treatment of T2D, the ensuing disaccharide maldigestion sometimes leads to intolerable gastrointestinal symptoms. The biological consequences of GLP-1 enhancement by α-glycosidase inhibition are also not characterised beyond their ability to minimise postprandial glucose fluctuations and delay in gastric emptying. Suppression of circulating NEFA by inhibition of adipose tissue hormone sensitive lipase may augment circulating GLP-1 concentrations.52 Drugs such as nicotinic acid, acipimox and to a lesser extent, fibrates, reduce circulating NEFA and are often employed in T2D for the treatment of dyslipidaemia, but are not well characterised in terms of possible incretin effects. A further approach would be to slow the degradation of the incretins by inhibiting DPPIV.
DIPEPTIDYL PEPTIDASE-IV INHIBITORS
Several DPPIV inhibitors (DPPIV-Is) are in clinical development; these agents have the advantage of being active in an oral form, with minimal gastrointestinal adverse effects (table 5). However, DPPIV enzymes metabolise a wide variety of peptides (e.g. peptide YY, neuropeptide Y, growth hormone-releasing hormone, vasoactive intestinal polypeptide) and therefore potentially affect other regulatory systems.53 – 55 Long-term safety data with DPPIV-Is are limited in patients with T2D, and both ongoing clinical trials and post-marketing surveillance are needed to clarify this issue. Of the two DPPIV-Is, vildagliptin and sitagliptin, sitagliptin has been approved for use in T2D in the USA at a dose of 100 or 200 mg daily, and vildagliptin, at a possible dose of 50 mg twice daily, is in late-stage development. Both vildagliptin and sitagliptin inhibit endogenous GLP-1 degradation, thereby relying on the ability to secrete GLP-1 (and GIP) in T2D.56 Since GLP-1 secretion may be defective, any benefit may be variable. This mechanism may explain the flat dose–response curve observed in trials of vildagliptin and sitagliptin. Vildagliptin increased B-cell mass in animal studies.57
The DPPIV-Is amplify the early insulin effect to glucose, and although fasting glucagon is not much altered, postprandial glucagon secretion is abolished.58 Data from trials suggest that the HbA1c is reduced by around 0.6%; the effect is sustained over time. Fasting plasma glucose levels are slightly reduced, and reductions in postprandial glycaemic measures are pronounced. Advantages of DPPIV-Is include oral administration, good tolerability, and weight neutrality. Gastrointestinal side effects are mild to moderate in severity, and although monotherapy in phase III trials showed no hypoglycaemia, rare hypoglycaemic events, especially in combination therapy, mostly mild, have been reported. Most data on DPPIV-I are derived from short-term clinical trials; the long-term safety of DPPIV inhibition must be evaluated in large trials.
INCRETIN ADMINISTRATION
GIP administration is ineffective in T2D and incretin replacement in reality refers to replacing GLP-1 bioactivity. This approach does not rely on adequate endogenous GLP-1 secretion and will be effective even in those with defective GLP-1 secretion. Although GLP-1 is effective in T2D, it has the disadvantage of requiring continuous intravenous infusion since it is degraded rapidly (⇑ tables 5 and 6). GLP-1-like agents that are resistant to DPPIV, such as liraglutide and exendin-4 (exenatide) may be given subcutaneously and intermittently.
Liraglutide (⇑tables 5 and 6)
A long-acting analogue of GLP-1 with 97% homology, with a palmitoyl fatty acid (C16) attached to a peptide, called liraglutide, has been produced (Novo Nordisk, Copenhagen, Denmark).59 The fatty-acyl-GLP-1 structure binds to interstitial albumin at the injection site. It is released slowly from the albumin complex and then absorbed into the circulation. The GLP-1–albumin complex results in a favourable pharmacokinetic profile because it resists DPPIV metabolism, delays absorption, and reduces renal clearance. The half-life of liraglutide is 12 hours, which allows for once-daily administration.60 The dose of liraglutide that produces clinically useful effects on HbA1c is between 0.45 and 0.75 mg daily and further clinical trials are needed to determine the optimal dose. Weight loss was also observed in clinical use.61 Nausea was the most common adverse effect, but most patients are able to continue using the drug. In combination therapy of T2D with liraglutide, hypoglycaemia occurred, but counter-regulatory hormone responses during hypoglycaemia were unimpaired.62 ,63 Long-term data are needed to fully assess its potential in treatment of T2D.
Exendin-4 (⇑tables 5 and 6)
Exendin-4, so named to describe an exocrine hormone (isolated from saliva of the Gila monster lizard, Heloderma suspectum) with endocrine properties, has been produced in synthetic form as exenatide (Byetta; Amylin Pharmaceuticals, San Diego, California, and Eli Lilly and Co., Indianapolis, Indiana, USA). It shares 53% amino acid sequence identity with human GLP-1.64 Exendin-4 peptide is DPPIV resistant, binds to the pancreatic GLP-1 receptor in vitro, and its pharmacological action lasts approximately 6 hours. After subcutaneous administration, exenatide reaches peak plasma concentrations in approximately 2 hours, its half-life is 2.4 hours, and it is predominantly eliminated by glomerular filtration.65 The dose of exenatide for licensed use in T2D in the USA is 5 or 10 μg subcutaneously twice daily. In clinical trials, exenatide lowered HbA1c values by approximately 1% compared with placebo, with substantial weight loss and lower fasting plasma glucose levels.66 – 68. Adverse effects were dose related, with nausea and vomiting being the most common. Titration to the target dose appeared to reduce the frequency of some adverse events.69 The risk of mild-to-moderate hypoglycaemia increased when exenatide was combined with a sulphonylurea but not when it was given with metformin. Exenatide, like liraglutide, produced hypoglycaemia, especially with combination therapy, but counter-regulatory responses to hypoglycaemia were unimpaired during hypoglycaemia.62 ,63 Approximately 40% of the patients exposed to exenatide developed low titres of antibodies. However, the anti-exenatide antibodies, in the main, did not affect patients’ clinical glycaemic control. About 6% of patients had high titres of antibodies; in about one half of these patients, the glycaemic response of exenatide appeared to diminish.65
Incretin mimetics such as liraglutide and exenatide, as well as DPPIV-Is, increased B-cell mass in animal studies57 and improved proinsulin/insulin ratio, first phase insulin and HOMA-B in humans, suggestive of improved B-cell function.70 – 74 The improvement in B-cell function and probable increase in B-cell mass with the incretin therapies may arrest the progression of B-cell failure in T2D and alter the natural history of the disease.
It is currently not clear at what point in the natural history of T2D incretin therapy should be instituted and more research is needed to address this issue. Logic dictates that since incretin failure may occur early and can address many pathophysiological features of T2D that lead to progression despite use of existing treatments, incretin therapy should be used early. However, most current data on incretin therapy are to be found in combination with existing treatments often when T2D is well advanced. Strategies to augment the biological actions of GLP-1 and/or GIP are expected to minimise weight gain, reduce hypoglycaemic episodes and prevent progressive B-cell failure by increasing B-cell mass. The optimal agent(s) that may mimic and replace the endogenous incretin effect is not fully known and awaits the outcome of ongoing clinical trials. Important issues with regard to practicalities such as oral or parenteral route of administration, long term safety and efficacy may determine which of the proposed options to increase the incretin effect is preferred in clinical practice.
Take-home messages
Gut hormones, insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1), are released in response to ingestion of carbohydrate-rich nutrients.
GIP and GLP-1, called incretins, augment the ability of the B-cell to secrete insulin and are crucial to maintaining euglycaemia.
Incretin failure, either failure of secretion or resistance to its action, is an increasingly recognised important factor in the hyperglycaemia of type 2 diabetes (T2D).
Incretins also produce weight loss, and induce a delay in gastric emptying, thus minimising postprandial glucose excursions and increasing B-cell number. These properties make them valuable agents in the treatment of T2D.
Replacement of incretin deficiency in T2D can be achieved by employing inhibitors of degradation of GIP and GLP-1 (DPPIV) or by parenteral administration of GLP-1 mimetic agents, liraglutide or exenatide. The exact place of these agents in treatment of T2D awaits clarification.
REFERENCES
Footnotes
Competing interests: None declared.