Article Text

Abstract
Background Treatment strategies for Crohn’s disease (CD) suppress diverse inflammatory pathways but many patients remain refractory to treatment. Autologous haematopoietic stem cell transplantation (SCT) is an emerging therapy for medically refractory CD though the mechanisms through which it circumvents refractory pathophysiology are unknown.
Objective The objective of this study is to understand how the immune system reconstitutes post-SCT and whether SCT may function as a cellular therapy restoring appropriately responsive immune cell populations from haematopoietic stem cells (HSCs).
Design Adults with CD with active clinical and endoscopic disease who failed available medical therapies were enrolled in a phase II study of SCT for refractory CD (n=19). Blood and intestinal samples were collected longitudinally and analysed using CyTOF and scRNA-seq. Stem cell autografts were functionally assayed in mouse xenograft models.
Results scRNA-seq and CyTOF analyses reveal that SCT predominantly affected the intestinal myeloid lineage with loss of inflammatory populations and return of macrophages capable of supporting mucosal healing. Xenograft models using patient HSCs suggested that HSCs support the early reconstitution of the myeloid lineage and reveal an impairment of short and long-term HSC engraftment that may determine SCT outcomes.
Conclusions This study suggests SCT functions as a myeloid-directed cellular therapy reinforcing the critical role of macrophages in refractory CD pathophysiology and as a target for cellular therapies. Furthermore, we report an unrecognised functional heterogeneity among HSC subpopulations in CD that may be relevant to our understanding of CD treatment and pathophysiology.
- AUTOIMMUNE DISEASE
- CROHN'S DISEASE
- STEM CELLS
- MACROPHAGES
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request. All data relevant to this study were created by the authors of this study. The scRNA-seq datasets have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE284677.1 All other data is available in this manuscript, supplemental materials, or will be made available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Despite diverse therapies available that target inflammatory pathways, many Crohn’s disease (CD) patients remain refractory to all available therapies leading to increased morbidity and mortality.
Stem cell transplantation (SCT) has emerged as a therapy for medically refractory CD.
No study has systemically explored how haematopoietic stem cells (HSCs) may drive immune reconstruction events that reverse refractory CD pathophysiology.
WHAT THIS STUDY ADDS
We demonstrate SCT acts as a myeloid-directed cellular therapy restoring intestinal macrophages capable of supporting mucosal healing.
Interrogation of HSCs in a xenograft model demonstrates functional impairment of HSCs in certain patients which may affect SCT outcomes.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study reinforces the central role of intestinal macrophages in CD biology and highlights the importance of understanding reparative cellular functions as drivers of refractory pathophysiology including growth factor signalling.
Functional impairment of HSCs in CD may contribute to refractory CD pathophysiology and impact CD treatments including SCT.
SCT is an important therapeutic option for medically refractory CD patients and must be considered as a cellular therapy to develop future protocols that improve safety and efficacy.
Introduction
Crohn’s disease (CD) is an immune disease that involves dysregulation of host-microbe interactions causing inflammation of the gastrointestinal tract.1 The standard treatment strategy for CD targets inflammatory pathways. Despite an increasing diversity of immunotherapeutic strategies, there remains a large number of patients who lose their initial therapeutic response increasing patient morbidity and mortality.1 The acquisition of a medically refractory phenotype appears independent of therapeutic mechanism as failure of any therapeutic modality leads to diminished efficacy with subsequent modalities. As such the development of medically refractory CD may be due to a redundancy in seemingly diverse immunotherapeutic strategies or that causes of medically refractory CD cannot be corrected by suppression of inflammatory pathways alone. Autologous haematopoietic stem cell transplantation (SCT) is an emerging therapeutic option for patients with medically refractory CD and other immune diseases.2 The clinical efficacy of SCT in the majority of medically refractory CD patients presents an opportunity to understand novel therapeutic pathways by which SCT reverses the processes underlying refractory CD.2–4
SCT was developed for the treatment of patients with cancer to facilitate the administration of high-dose chemotherapy by using haematopoietic stem cells (HSCs) to prevent chemotherapy-induced myelosuppression. The mechanisms by which SCT results in a therapeutic benefit for refractory CD and other immune diseases are unknown. When SCT was first explored as a treatment for immune diseases it was believed that SCT may similarly allow for administration of high-dose chemotherapy to suppress the aberrant inflammatory response. Our hypothesis is that HSCs in SCT protocol may function more as a cellular therapy restoring appropriately responsive immune cell populations from haematopoietic progenitors in the stem cell autograft (ie, an immune ‘reset’).2 Studies in patients with cancer have examined immune reconstitution following SCT but have not systematically explored such events as they might be relevant to SCT as a cellular therapy including how HSC subpopulations re-establish cellular immunity in tissue niches.5 6 Initial studies to understand mechanisms by which SCT alleviates the consequences of medically refractory CD profiled circulating immune cells using flow cytometry, but these studies did not shed light on how HSCs drive immune reconstitution events in the intestine.5 7 Here we aim to advance our hypothesis that SCT may act as a cellular therapy for CD and report how HSCs determine immune reconstitution events in CD using high-dimensional cellular phenotyping and functional studies of the stem cell grafts. Using single-cell RNA sequencing (ie, scRNA-seq) and mass cytometry (ie, CyTOF) we have characterised immunophenotypic changes in the peripheral blood and the intestine of recipient patients that indicate SCT primarily acts as a myeloid-directed cellular therapy restoring intestinal macrophages capable of supporting mucosal healing. These studies highlight the central role of monocytes/macrophages in driving CD pathophysiology and propose the acquisition of medically refractory CD may not be secondary to unknown inflammatory pathways but rather the impairment of pathways supporting mucosal healing. Functional studies of stem cell grafts in a xenograft model further revealed a previously unrecognised heterogeneity in the composition and function of mobilised HSCs which may determine these immune reconstitution events, SCT efficacy and suggest that CD pathophysiology extends to the marrow niche.
Materials and methods
See online supplemental materials.
Supplemental material
Patient and public involvement
No patients or public were involved in the design of this study.
Results
SCT is highly effective in refractory CD patients
Patients with medically refractory CD were enrolled in the MASCT-CD trial. 19 patients were enrolled from 2018 to 2023 and 14 were followed for at least 6 months of follow-up post-SCT (figure 1A). At 6 months post-transplant 10/14 patients had an endoscopic remission (total SES-CD≤4 and no sub-score>1), 13/14 had an endoscopic response (reduction of SES-CD by at least 50%) and 8/14 patients had histological healing (normalisation or inactive chronic) (figure 1B–E).8 9 Paired blood and intestinal tissue samples were obtained at baseline and 6 months post-SCT and additional blood samples were taken longitudinally (figure 1F).

SCT is highly effective in refractory CD patients. (A) Table of patient demographics at baseline. (B) Bar graphs of clinical outcomes at 6 months and 12 months post-SCT demonstrate a significant reduction in Harvey Bradshaw Index (HBI) and Crohn’s Disease Activity Index (CDAI) scores (n=14, mean±SEM). (C) Line graph of paired samples for endoscopic outcomes using Simple Endoscopic Score for Crohn’s Disease (SES-CD) at 6 months post-SCT (n=14). (D) Pie graphs of endoscopic response (SES-CD decrease ≥50%) and endoscopic remission (SES-CD≤4 and no subscore >1 in any individual variable) at 6 months post-SCT. All statistical analysis (B and C) was done using paired Wilcoxon signed-rank test with *p<0.05, **p<0.01 ***p<0.001 ****p<0.0001. (E) Bar and pie graph of subjects with all intestinal biopsies at 6 months post-SCT that were histologically normal or inactive with only chronic changes (n=14). (F) Schematic of MASCT-CD clinical trial and sample collection timeline. CD, Crohn’s disease; SCT, stem cell transplantation.
SCT has a distinct effect on intestinal immune populations
To characterise immune reconstitution events following SCT, blood and intestinal samples were analysed longitudinally by CyTOF. Supervised clustering of immune cell populations was performed using canonical markers and unsupervised clustering using FlowSOM.10 11 Iterative FlowSOM analyses performed independently on intestinal and blood samples organised immune cells into 15 metaclusters (ie, parental population) inclusive of 100 cell clusters (ie, sub-population). Metaclusters demonstrated concordance with supervised immune cell populations defined by canonical protein markers (online supplemental data 1). All samples were taken from patients with an endoscopic response to SCT (figure 1).
Supplemental material
The effect of SCT on immune cell populations 6 months post-SCT suggested distinct changes to the circulating and the intestinal immune cell compartments (figure 2A, online supplemental figure 1). Analysis of FlowSOM organised metaclusters identified significant changes to circulating lymphoid populations and intestinal myeloid populations reinforcing the specific effect of the transplant on each immune compartment. Analysis of metacluster sub-populations (ie, clusters) indicated far greater changes to intestinal immune cell populations with significant changes in 21/100 immune cell clusters compared with circulating immune cell clusters (3/100 clusters) (figure 2B). A principal component analysis of FlowSOM organised immune cell clusters supported an effect of SCT on intestinal immune cell populations (figure 2B). Supervised and unsupervised analyses demonstrated that the most significant changes post-SCT involved circulating CD27− B cells (metacluster 3) and intestinal CD14+ cells (metacluster 1) (figure 2C, online supplemental figure 1). Prior studies of immune reconstitution events post-SCT in patients with CD similarly reported an effect on circulating naïve B cell populations.5 Within intestinal metacluster 1 there were significant increases post-SCT in three clusters that varied in expression of CD14, CD206, CD4, CD64, CD11c and HLADR confirming monocyte-derived cell populations. Metacluster 1 clusters were CD11clo CD86lo (cluster 24, 33) and CD14lo CD206lo (cluster 24) consistent with developing homeostatic macrophages.12 13 Metacluster 1 cluster 2 expressed CD141hi CD1clo CD103lo consistent with monocyte-derived DC. The most significant increase in metacluster 1 CD14+ populations was in patients with histological normalisation post-SCT whereas patients with histological inflammation had persistent suppression of CD14+ populations (online supplemental figure 1D). Our findings reveal a distinct and significant effect of SCT on intestinal immune cell populations highlighting the importance of defining immune reconstitution events in different niches including blood and intestine.

Stem cell transplantation (SCT) has a distinct effect on intestinal immune populations. (A) Unsupervised CyTOF analysis with FlowSOM for blood (top) (baseline n=7, 6 months post-SCT n=9) and intestinal (bottom) (baseline n=13, 6 months post-SCT n=13) samples with each minimal spanning tree diagram displaying 15 metaclusters comprised of 100 clusters. Metaclusters are annotated using canonical cell surface markers or prominent markers if lineage markers are negative (lin- are CD3−CD19−CD14−CD16−CD56−CD66b−). Metaclusters that were significantly changed from baseline to 6 months post-SCT are indicated by red font and red dashed circle and clusters that were significantly changed are indicated by solid red circle (p<0.05, Mann-Whitney test). (B) Bar graph depicts the number of significantly changed clusters from baseline to 6 months post-SCT (p<0.05, Mann-Whitney test). Principal component analysis (PCA) of blood (top) and intestine (bottom) samples comparing baseline with 6 months post-SCT (FlowSOM clusters, PCA analysis 95% CI). (C) Bar graph (mean±SEM) of most significantly changed cell populations in supervised clustering analyses from blood (baseline n=9, 6 months post-SCT n=11) and intestine (baseline n=13, 6 months post-SCT n=13), Mann-Whitney test, ***p<0.001, ****p<0.0001.
Reconstitution of intestinal myeloid populations
To further understand how immune sub-populations are collectively reconstituted post-SCT, CyTOF datasets were analysed again by CITRUS.14 CITRUS organises single cells into hierarchical clusters that predict how a system responds to a perturbation (eg, SCT). CITRUS analysis confirmed that a larger number of intestinal immune cell clusters were affected by SCT compared with circulating immune cell clusters (figure 3A,B). Among circulating immune cells there was a significant increase post-SCT in a single cluster network including three related transitional B cell clusters that are CD19+CD27−CD38+BTLA+ (figure 3A, online supplemental figure 2).15 16 These transitional B cell clusters expressed HLADR, CD45RA, CD1c, CCR6 and integrins β7 and CD49d (α4) suggesting intestinal migrating transitional B cell populations including memory cells.17 Parental cell clusters to the B cell clusters did not express canonical lineage markers suggesting that they were progenitor cell populations. These progenitor cell clusters expressed integrins CD62L, CD29 and CD49d(α4) consistent with their potential to participate in intestinal trafficking.18

Reconstitution of intestinal myeloid populations. (A) CITRUS analysis of CyTOF datasets from blood (baseline n=9, 6 months post-SCT n=11) and (B) intestinal (baseline n=13, 6 months post-stem cell transplantation (post-SCT) n=13) samples. Hierarchical clusters are depicted as a CITRUS tree with each node representing a different cluster coloured coded by immune cell populations identified through expression of canonical protein markers. Cluster networks (>1 cluster) that collectively predict the change in immune cell populations 6 months post-SCT compared with baseline are outlined in red (significance analysis of microarrays, false discovery rate <0.01). Hierarchical relationships between significant clusters are identified with red arrows indicating the direction from parental to child cluster. (C) CyTOF samples were concatenated at each timepoint and CD14+ populations were analysed by viSNE. Overlapping tSNE plots demonstrate a change in intestinal CD14+ cell phenotypes between baseline and 6 months post-SCT.
CITRUS analysis of the intestinal immune compartment identified significant changes post-SCT to three large cell networks (figure 3B). These three cell networks included B cells, neutrophils and CD14+ cells (online supplemental figure 3).16 In contrast to the circulating B cell network there are a small number of CD19+CD27+CD38+ intestinal memory B cell clusters which were significantly decreased post-SCT. A neutrophil cell network was also significantly reduced post-transplantation. These neutrophil clusters expressed traditional markers including CD66b, CD11c, CD33, CD172a and CD11b.19 Neutrophil cell clusters that were significantly decreased post-SCT were distinct from neutrophil clusters unaffected by SCT based on their expression of CD69 suggesting a specific loss post-SCT of activated neutrophils.20 The CD14+ cell cluster network significantly increased post-SCT. This CD14+ cell cluster network expressed macrophage markers including HLADR, CD33, CD206, CD64, CD11b, CD11c and CD4.13 Within this CD14+ cell network were clusters that variably expressed CD206, CD11b and CD11c delineating changes to early differentiating monocytes/macrophages (CD11chi) as well as mature macrophage populations (CD206hi CD11blo).13 For each major myeloid population (eg, neutrophil, CD14+ cells) there was a significant change in the parental cluster for the lineage suggesting that the SCT perturbs intestinal myeloid lineages (figure 3B). There was also a significant increase post-SCT in a parental CD3+ T cell cluster at a branch point in the CITRUS tree between tissue-resident (CD103+) and effector (CD103−) populations. The parental CD3+ T cell cluster does not express naïve T cell markers including CD25 or CD45RA suggesting post-SCT there is an expansion of memory effector cell populations and no change to tissue-resident populations. To visualise global effects of SCT on major intestinal cell lineages we performed a viSNE analysis of manually gated T cell, B cell, neutrophil and CD14+ cell populations. This analysis confirmed the dramatic effects of SCT on intestinal myeloid populations with the almost complete phenotypic remodelling of CD14+ cells consistent with reconstitution of the myeloid lineage post-SCT (figure 3C, online supplemental figure 4).
SCT restores homeostatic macrophages in refractory CD patients
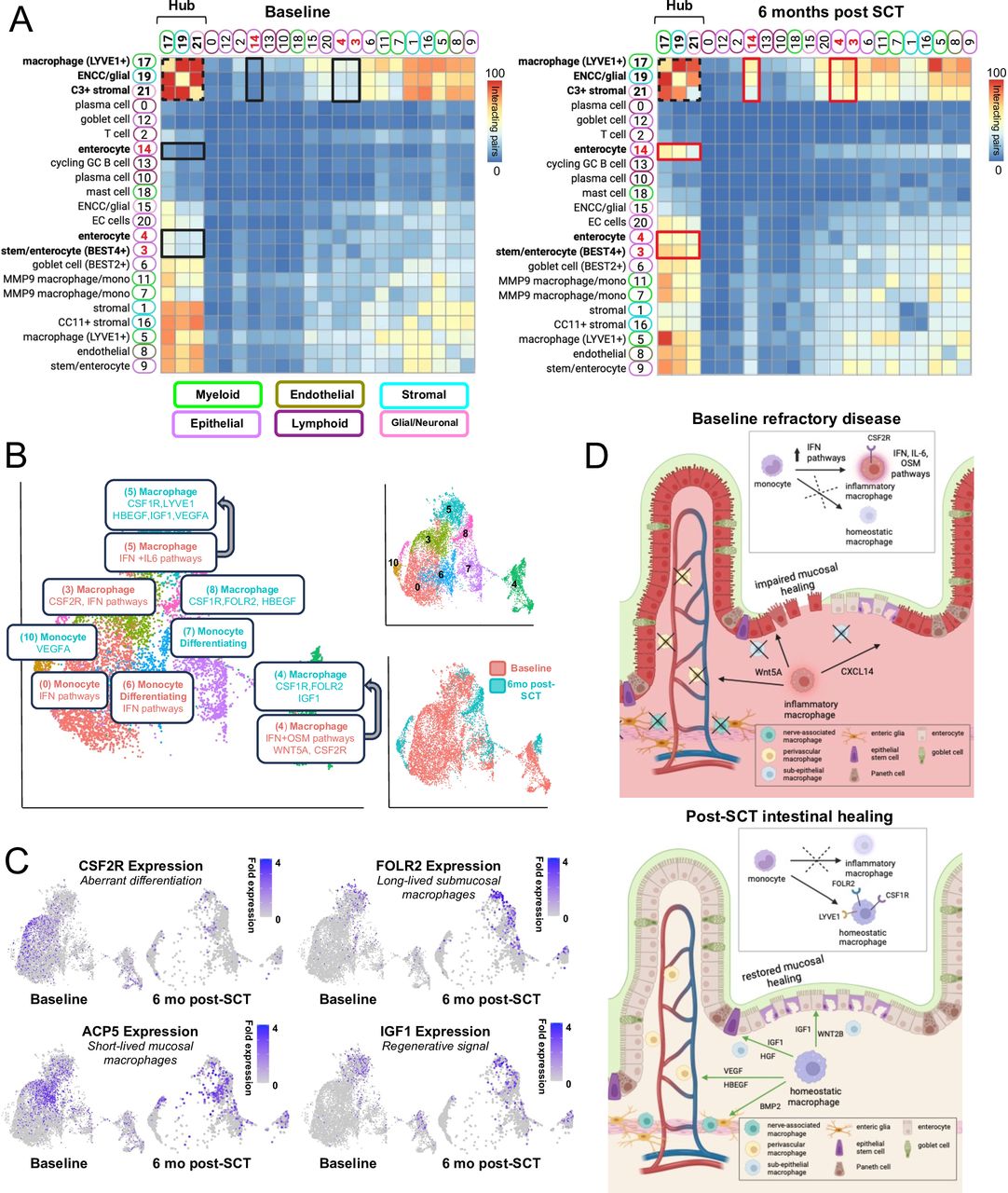
Proteomic (ie, CyTOF) studies suggested a disproportionate effect of SCT on intestinal myeloid populations. To further explore functional changes among intestinal cell populations, samples at baseline and 6 months post-SCT were analysed with scRNA-seq (data available at NCBI GEO database accession GSE284677).21 Single cells were clustered and annotated using known sets of marker genes and reference data sets resolving major cell population clusters (online supplemental figure 5, online supplemental data 2).22 All samples were taken from patients with an endoscopic response to SCT. Clustered scRNA-seq data were analysed by CellphoneDB to define ligand-receptor pairs that comprised cellular interaction networks (figure 4A).23 Among cell clusters, the stromal, myeloid, endothelial and glial cell clusters were predicted to interact extensively in the intestine whereas lymphoid clusters (B cell, T cell) had the smallest predicted interaction networks. CellphoneDB identified the strongest interaction network involving myeloid cluster 17 (macrophage/LYVE1), stromal cluster 19 (C3+ stromal) and glial cluster 21 (ENCC glial) (ie, hub clusters) (figure 4A). Hub clusters interacted broadly with other intestinal cell clusters confirming the importance of myeloid, stromal and glial cells as regulators of intestinal homeostasis.24 25
Supplemental material

SCT restores homeostatic macrophages in refractory CD. (A) scRNA-seq datasets from the intestine were analysed by CellphoneDB (baseline n=4, 6 months post-SCT n=3). Significant clusters pairing expression of ligand-receptor pairs displayed as a heatmap. Clusters with the largest number of interacting pairs at baseline and follow-up are outlined in a dashed black square (ie, hub clusters). The largest increase in interactions from baseline to 6 months post-SCT is between hub clusters and epithelial clusters 3, 4, 14 outlined in black at baseline and red at 6 months post-SCT. (B) UMAP plots from scRNA-seq intestinal dataset with re-clustered myeloid cells annotated (baseline n=4, 6 months post-SCT n=3). Clusters are highlighted based on abundance at each timepoint with clusters common to both timepoints that undergo remodelling indicated by arrows (online supplemental figure 5). Selected significant pathways and marker genes are highlighted for each cluster including DEGs for clusters common to both timepoints. (C) UMAPs demonstrating fold expression of select genes. (D) Schematic of monocyte/macrophage populations and functions that reinforce refractory CD pathophysiology at baseline or intestinal healing post-SCT. CD, Crohn’s disease; SCT, stem cell transplantation.
When comparing interactions in samples at baseline compared with 6 months post-SCT the most notable increase occurred between hub clusters 17, 19 and 21 involving epithelial clusters 3 (stem/enterocyte BEST4+), 4 (enterocyte) and 14 (enterocyte) (figure 4A). Examination of ligand-receptor pairs revealed signals involving chemokines, extracellular matrix, growth factors and WNT pathway ligands. At baseline, we observed limited interactions predicted to impair epithelial migration and wound healing, while post-transplant we observed large numbers of interactions predicted to support epithelial homeostasis and regeneration. At baseline cellular interactions that impaired epithelial migration, stem cell differentiation and wound healing involved CXCL14/CXCR4 and WNT5A/PTPRK.25–27 After SCT there was a complete loss of CXCR4/CXCL14 and WNT5A/PTPRK interactions with a striking increase in interactions involving diverse growth factors including VEGFA, IGF1, HBEGF and HGF that support epithelial stem cell maintenance, epithelial differentiation and epithelial growth.28 29 Growth factor signalling extended broadly across myeloid, stromal, glial and epithelial interaction networks post-SCT confirming a specific role for myeloid cells within the immune system to regulate intestinal homeostasis (online supplemental figure 5).12 24 30 31
To further understand functional changes among intestinal myeloid cells post-SCT, clusters 5, 7, 11 and 17 were re-clustered and annotated (figure 4B). As in the CyTOF analysis there was extensive remodelling post-SCT of intestinal monocyte and macrophage populations suggesting reconstitution of this immune lineage (figure 4B inset). Similarly, increased expression of CD14, CD206, CD141 and decreased expression of CD86 and CD11c post-SCT in the scRNA-seq dataset paralleled observations in the CyTOF dataset with significant increases in CD14+CD206+CD86loCD11clo and CD14+CD141+ populations (online supplemental figures 1 and 5). While certain monocyte/macrophage clusters appeared to be gained or lost post-SCT others were phenotypically remodelled (figure 4B, online supplemental figure 5). Pathway analyses and examination of signalling molecules revealed a lineage shift post-SCT from short-lived inflammatory mucosal populations with altered differentiation pathways towards long-lived homeostatic populations with normal differentiation pathways (figure 4C, online supplemental figure 5). Pre-SCT monocyte and macrophage clusters were enriched for interferon signalling pathways with certain macrophage clusters expressing oncostatin M (cluster 4) and IL-6 (cluster 5) signalling pathways associated with medically refractory CD patients.30 32 Cluster 3 and 4 macrophages pre-SCT expressed CSF2RB which promotes aberrant differentiation of inflammatory macrophage populations with impaired restorative functions.12 32 Post-SCT there was significantly increased expression of genes across the macrophage lineage suggesting differentiation of long-lived homeostatic tissue-resident cells expressing restorative signalling molecules (figure 4B,C). There was also a significant decrease post-SCT in the expression of genes in the type II interferon signalling pathway associated with the resolution of inflammation and loss of inflammatory macrophage populations. Homeostatic macrophage populations expressed diverse signalling molecules consistent with subepithelial macrophages that maintain epithelial integrity (WNT2B, HGF), perivascular macrophages that support angiogenesis (LYVE1, VEGF) and nerve-associated macrophages that maintain enteric nerves (BMP2) (figure 4B–D, online supplemental figure 5).12 Among signalling molecules the expression of IGF1 was a marker of macrophage populations enriched post-SCT (figure 4C). IGF1 and insulin signalling pathways are critical to intestinal healing suggesting absence of these functions in macrophages pre-SCT may impair intestinal healing in medically refractory CD.33 34
scRNA-seq analyses confirmed previous studies that suggested differentiation of aberrant macrophage populations reinforce an inflammatory intestinal niche in medically refractory CD.12 31 Our analyses further suggest aberrant macrophage differentiation may lead to loss of restorative functions including growth factor signalling required to coordinate cellular networks necessary for mucosal healing (figure 4D). Analysis of changes in the frequency of intestinal immune cell clusters confirmed the greatest change post-SCT was the loss of inflammatory MMP9 macrophages and the greatest increase in cells from a cycling GC B cell cluster and a T cell cluster (online supplemental figure 5). The previous CyTOF analysis did not suggest a similar increase post-SCT in mucosal B cell populations but did identify an increase in effector T cells (online supplemental figure 1, figure 3). Analysis of transcriptional changes in the T cell cluster post-SCT identified a significant increase in nuclear receptor pathways associated with ZNF683 consistent with expansion of tissue-resident memory effector T cell populations.35 36 The expansion of cycling GC B cells and T memory effector cells supports that mucosal lymphocyte changes post-SCT may be characterised by the expansion of resident memory populations and not an influx of naïve populations. Analysis of changes in the frequency of non-immune cell clusters confirms a large increase in cells from enterocyte cluster 4 and C3+ stromal cluster 19 whose interaction networks were observed to increase post-SCT (figure 4A). C3+ stromal cells are important to myeloid differentiation and epithelial barrier functions suggesting that restoration of these stromal-myeloid interactions post-SCT may be necessary to further support epithelial healing.37 38
SCT is a myeloid-directed cellular therapy
scRNA-seq and proteomic profiling of immune reconstitution events post-SCT suggested remodelling of intestinal myeloid populations consistent with immune cell reconstitution of this lineage. The ability of SCT to reconstitute (ie, reset) an immune lineage and function as a cellular therapy requires replacement of resident immune populations from progenitors within the stem cell graft. To characterise early immune reconstitution events post-SCT and interrogate the relationship between immune reconstitution events with progenitor populations we performed scRNA-seq of blood samples at baseline, stem cell collection, engraftment, 3 months post-SCT and 6 months post-SCT.
Cell clusters specific to stem cell collection or engraftment were identified and could not be annotated using gene markers for mature immune cell subsets suggesting that they represent progenitor populations (figure 5A, online supplemental figure 6). Three cell clusters (21, 24, 25) specific to the time of stem cell collection and engraftment expressed CD34 consistent with their being haematopoietic progenitor cells or HSCs (online supplemental figure 6). To establish ontological relationships between potential progenitor cell clusters and mature immune cell clusters we analysed scRNA-seq datasets using scVelo (figure 5A).39 scVelo quantifies the ratio of spliced to unspliced RNA (ie, RNA velocity) to predict ontological relationships between cell populations. RNA velocity analysis confirmed the majority of clusters specific to stem cell collections and engraftment were lineage-committed myeloid progenitor populations with relationships to mature circulating myeloid cells. To further understand HSC populations, clusters 21, 24 and 25 were re-clustered and annotated. Re-clustering of CD34+ populations distinguished populations specific to stem cell collection (2, 4, 7), engraftment (0, 1, 3, 6) and both timepoints (5, 8) (figure 5B). All CD34+ cell clusters were annotated as bone marrow cells confirming the relationship of mobilised and marrow-resident HSC populations. CD34+ cell clusters included HSC multipotent progenitors (HSC-MPP), cycling HSC-MPP, common myeloid progenitors, megakaryocyte-erythroid progenitors and granulocyte-monocyte progenitors (figure 5B). Lymphoid-committed CD34+ cell clusters were not observed suggesting a bias in mobilised HSCs towards myeloid differentiation.

SCT is a myeloid-directed cellular therapy. (A) Blood scRNA-seq samples from all timepoints (n=20) were clustered with UMAP plots highlighting progenitor clusters specific to stem cell collection (n=4) and engraftment (n=3) that are not found at baseline (n=7). RNA velocity curves using scVelo of stem cell collection demonstrate progenitor clusters related to mature myeloid cells. (B) CD34 expressing clusters were re-clustered and annotated. Clusters are highlighted based on their presence at engraftment or stem cell collection. (C) Bar graph (mean±SEM) of Simpson clonality of TcRβ sequencing of blood (baseline n=10, 6 months post-SCT n=10) and tissue (baseline n=9, 6 months post-SCT n=9) samples at baseline and 6 months post-SCT, statistical analysis with Dunn’s test, *p<0.05. (D) Bar graph (mean±SEM) of max clone frequency for T cell clones present at both timepoints, statistical analysis with Dunn’s test, *p<0.05. The increase in Simpson’s clonality post-SCT in the blood and intestine suggests a decrease in T cell clonal diversity. HSC, haematopoietic stem cell; SCT, stem cell transplantation.
scRNA-seq analyses suggest that SCT may act as a myeloid-directed cellular therapy driving the early expansion of myeloid progenitors capable of reconstituting intestinal myeloid populations. Analysis of CyTOF datasets supports the scRNA-seq analyses and identified lineage-negative populations at engraftment that express myeloid markers and integrins necessary for intestinal trafficking (online supplemental figure 6). To further assess the impact of SCT on lymphoid populations we analysed changes in T cell clonality through analysis of T cell receptor (TCR) diversity. TCRβ sequencing demonstrated a significant increase post-SCT in T cell clonality in circulating and intestinal T cell population and a trend towards a decrease in T cell clone diversity post-SCT in the intestine (figure 5C, online supplemental figure 7). Analysis of clones present at each timepoint confirmed an increase post-SCT in the frequency of these clones suggesting the persistence and expansion of T cell clones post-SCT (figure 5D). Among the highest-frequency clonal populations many clones remained at a high-frequency post-SCT, especially in the intestine (online supplemental figure 7). These findings further support that the current SCT protocol may selectively target intestinal myeloid populations and is insufficient to fully reconstitute T cell populations which may be replenished post-SCT through clonal expansion of persistent memory populations and not by progenitors in the stem cell graft.40 41
HSC heterogeneity in CD
To further interrogate HSC populations and their contribution to immune reconstitution post-SCT, we performed functional and phenotypic analyses of the stem cell grafts that were administered to CD patients. Cryopreserved mononuclear cell grafts were obtained from the Mount Sinai Blood Bank and thawed per clinical protocol. Patient samples contained an average of 4.23% CD34+ HSCs (range 2.1%–8.49%). Phenotypic analysis of CD34+ cell populations was performed by flow cytometry to delineate short-term (ST), intermediate-term (IT) and long-term (LT) HSCs and lineage-committed progenitors (figure 6A, online supplemental figures 8 and 9). The composition of these CD34+ populations varied across patient samples with the predominant populations being ST-HSC or lineage committed progenitors consistent with the scRNA-seq analyses.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HSC heterogeneity in CD. (A) Flow cytometry analysis of mobilised stem cells from nine patients. Stem cell populations are percent live CD34+ cells. (B) Human CD34+ stem cells were injected into NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice with mean line graphs of CD45+ populations in the bone marrow of the mice from three patients with the highest engraftment (as shown in figure 6C) for granulocytes (CD15+), monocytes (CD14+), B cells (CD19+), T cells (CD3+), myeloid cells (CD33+) and megakaryocytes (CD41a+) (mean, n=2 biological replicates at each timepoint). (C) Analysis of engrafted human CD45+ cells in the murine bone marrow at 4, 8 and 16 weeks. Bar graph of % human cells from all cells isolated (mean, n=2 biological replicates at each timepoint). Pt ID key identifies clinical response for each patient. Graph background colour demonstrates the contribution of ST-HSC and LT-HSC to human engraftment at each timepoint. CD, Crohn’s disease; HSC, haematopoietic stem cell; IT-HSC, intermediate HSC; LT-HSC, long-term HSC; MPP, multipotent progenitors; SCT, stem cell transplantation; ST-HSC, short-term HSC.
To functionally interrogate the HSC grafts, their CD34+ cells were transplanted into NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice.42 HSC engraftment and cell differentiation potential was assessed at weeks 4, 8 and 16 within the bone marrow, spleen and peripheral blood of recipient mice using flow cytometry (figure 6B,C and online supplemental figures 8 and 9). Examination of the human cell subpopulations confirmed the presence of functional HSCs capable of multilineage hematopoiesis (figure 6B, online supplemental figure 8). Analysis of mature CD45+CD34− cells at week 4 indicated that CD33+ myeloid cells were the predominant cell population in the bone marrow. At week 8 there was an increase in CD19+ cells which decreased by week 16 with CD33+ cells again being the dominant population. Mature CD3+ T cells were detected only transiently and at exceedingly low levels in all mice including analysis of the spleen and blood (online supplemental figure 8).43 The dynamics of immune reconstitution events in the xenograft model paralleled patient observations where myeloid progenitors emerged at engraftment followed by a rise in naïve B cell populations and a persistent decrease in naïve T cell populations (online supplemental figure 4).
At each timepoint, the engraftment of human CD45+ cells in the bone marrow of recipient mice ranged from 0% to 71% (figure 6C). The greatest degree of engraftment was observed at week 8 which was reduced by week 16. Engraftment of human CD34+ cells in the bone marrow was similarly reduced by week 16 likely reflecting the limited number of functional LT-HSCs present in these grafts (online supplemental figure 8).44 The observed functional heterogeneity of HSC populations in the xenograft model despite classification using established protein markers suggests dysfunction in HSC differentiation and repopulation capacity especially among LT-HSC.43 45 Correlation between HSC function in the xenograft model and SCT clinical outcomes is not feasible due to the rarity of clinical non-response in this study at 6 months (1/14 patients). In follow-up to 12 months a second patient had a clinical/endoscopic relapse 8 months post-SCT. It was observed that stem cell grafts from the two patients with a negative SCT clinical outcome failed to engraft in the xenograft model whereas stem cell grafts from four patients with stable endoscopic and clinical outcomes post-SCT achieved human cell chimerism in the xenograft model (figure 6C). The functional heterogeneity of HSC from CD patients in the xenograft model is inconsistent with historical observations in healthy patients suggesting a systemic impact of CD on HSC populations.43–45
Discussion
Our observations suggest that SCT functions as a myeloid-directed cellular therapy in CD involving HSC-mediated reconstitution of intestinal macrophages capable of supporting mucosal healing. These observations are independently validated using multimodal studies of immune reconstitution events including CyTOF and scRNA-seq. High-dimensional cell profiling methods capture cellular diversity but are limited by the lack of functionally annotated reference datasets and the inability to resolve certain cell populations such as neutrophils with scRNA-seq. Future functional studies of scRNA-seq or CyTOF-resolved cell clusters will be critical to validate how specific myeloid populations impair or reinforce mucosal healing. The functional studies presented here of HSCs in xenograft models paralleled clinical observations in patients including the early reconstitution of the myeloid lineage, impaired reconstitution of T cell populations, and potentially predicted the clinical efficacy of SCT in patients with functional engrafting HSC.
The observations in the xenograft models that graft-mediated reconstitution of the marrow niche may be a critical therapeutic outcome suggests the inability to repopulate HSC populations in the marrow may underlie the consistent clinical observation that SCT is incapable of maintaining long-term disease-free remission in many patients.3 4 46 47 A limitation of this study is the lack of patients without an endoscopic response to facilitate an understanding of how specific HSC populations or immune reconstitution events predict treatment non-response. Future studies in larger cohorts over longer periods of time that capture events related to treatment non-response will be critical to validate the therapeutic mechanisms proposed in this study and whether functional HSC heterogeneity determines SCT outcomes. The heterogeneity of HSC populations in CD as determined by functional analyses of the stem cell grafts further suggests CD pathophysiology may create a dysfunctional marrow niche shaped by the CD inflammatory milieu. The functional impairment in xenograft models of ST-HSC and LT-HSC populations is consistent with studies that demonstrate an impact of chronic inflammation on these HSC populations.48 49 The importance of how CD affects the function of specific HSC populations likely extends beyond therapeutic implications proposed here and may be critical to how HSCs reinforce disease pathophysiology, especially in the myeloid lineage. The studies presented here suggest a hypothesis wherein the pathophysiology of refractory CD is acquired through changes to haematopoietic progenitors in the marrow from chronic inflammation leading to sustained aberrant myeloid differentiation.46 47 This hypothesis provides a plausible explanation for why a cellular therapy like SCT is necessary to circumvent refractory CD pathophysiology by restoring healthy progenitor populations capable of normal myeloid differentiation (online supplemental figure 10).46 47
The successful use of SCT as a cellular therapy requires an understanding of how haematopoietic progenitors shape tissue-resident immune populations. The intestinal myeloid niche is largely dependent on circulating progenitor populations and as such may be more amenable to reconstitution with SCT.12 13 The inability of the current SCT protocol to adequately reconstitute the intestinal lymphoid lineages, especially T cells, may be secondary to the observed myeloid bias of the harvested stem cell grafts, age-related changes in hematopoiesis and limitations of the conditioning chemotherapy to deplete tissue resident lymphoid memory cell populations.40 50 These findings suggest that future SCT protocols may improve clinical efficacy through plerixafor-based mobilisation strategies that enrich healthy long-term progenitors to facilitate complete immune reconstitution.51–53 While the persistence of tissue-resident lymphoid memory populations did not impact short-term clinical outcomes, it is possible that re-expansion of persistent pathogenic clones may contribute to the high rates of long-term clinical relapse post-SCT. While future studies will be needed to link specific immune reconstitution events to long-term clinical outcomes, the data presented here suggests additional lymphodepletion with ATG may not be necessary and would improve the safety of conditioning protocols. Patients in this study were exposed to vedolizumab after immune reconstitution requiring additional studies to deconvolute the effect of vedolizumab from our observations. A previous study of SCT in CD patients who were not exposed to vedolizumab reported similar changes in the circulating B cell compartment though did not directly interrogate immune reconstitution events in the intestine.5 A recent study of ulcerative colitis patients exposed to vedolizumab observed a decrease in intestinal CD14+ populations suggesting the observed changes in CD14+ populations post-SCT may not be secondary to vedolizumab alone. With a greater understanding of the interplay between SCT conditioning and mobilisation regimens with HSC-mediated immune reconstitution events, we anticipate next-generation SCT protocols will safely and efficiently target pathogenic immune cell lineages and sustain immune reconstitution events over time.
The last two decades have brought unprecedented growth in CD therapies, but it has remained consistent that each new therapy is less effective for patients that failed a previous therapy.8 54 While CD therapies seemingly encompass distinct therapeutic mechanisms, all therapies target the inflammatory response supporting the concept that medically refractory disease involves pathways not directly related to inflammation. This study suggests SCT acts as myeloid-directed cellular therapy restoring macrophage functions critical to intestinal healing. That the pathophysiology of medically refractory disease involves intrinsic defects in the differentiation of cell populations required to heal the intestine after resolution of inflammation provides a plausible hypothesis for why therapies that target inflammation alone fail in medically refractory patients.1 9 The findings in this study further reinforce the central role of intestinal macrophages in CD biology including medically refractory disease.12 30–32 Another factor that might contribute to the recurrence of CD after SCT is the inability of autologous HSCs to correct genetically driven defects in intestinal macrophages.55 The studies presented here suggest the genetic correction of HSCs in SCT grafts may be sufficient to restore wild-type function of resident myeloid cells in the intestine of CD patients leading to long-term disease-free remission.56 While the widespread use of SCT in CD patients is limited by toxicity, this study adds to the growing literature confirming the clinical efficacy of SCT for patients with medically refractory CD. The therapeutic applications of observations made in this study of SCT may be generalisable to all patients with CD through the continued development of regenerative and cellular therapies that support tissue healing as a novel strategy to complement the abundance of existing therapies that suppress inflammation.57–60
Supplemental material
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request. All data relevant to this study were created by the authors of this study. The scRNA-seq datasets have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE284677.1 All other data is available in this manuscript, supplemental materials, or will be made available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Mount Sinai IRB Study #16-01304. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
At the Icahn School of Medicine at Mount Sinai we acknowledge the Human Immune Monitoring Center for assistance in the CyTOF assays. This work was supported by T32 grant 1T32DK131953-01A1. This work was supported in part through the computational and data resources and staff expertise provided by Scientific Computing and Data at the Icahn School of Medicine at Mount Sinai and supported by the Clinical and Translational Science Awards (CTSA) grant UL1TR004419 from the National Center for Advancing Translational Sciences. We would like to acknowledge Dr Camelia Iancu-Rubin and Dr Suzanne A Arinsburg from the Cellular Therapy Laboratory at Mount Sinai for their assistance in acquiring and processing peripheral mobilised blood stem cell collections. Samples were acquired using Cellular Therapy Protocol HS#21-00625. We would like to acknowledge the MASCT-CD study team; Alema Gonzalez, Ladislao Decenteceo, Jonathan Lagdameo, Moutasem Mansi, Stephanie Gold, David Faleck, Amir Steinberg, Saurabh Mehandru, Bruce Sands, Ryan Ungaro, Alexander Greenstein, Laurie Keefer and Laura Manning. We thank the Duke University School of Medicine for the use of the Sequencing and Genomic Technologies Shared Resource, which provided sequencing service for scRNA-seq samples. All graphical illustrations were created with BioRender.com.
References
Footnotes
X @Felix_Chuang240
Contributors Guarantor: LC. Conceptualisation: LC. Methodology: LC, J-FC, JC, RH. Software: ST, CT. Validation: all authors. Formal analysis: DG, ST, XW, LC. Investigation: DG, ST, XW, AD, AE, EF, CL-S, KS, SS. Resources: XW, AD, AE, J-FC, CT, JEL, JLMF, RH, JC, LC. Data curation: DG, ST, EF, SS, LJC. Writing – original draft: DG, LC. Writing – review and editing: all authors. Visualisation: DG, LC. Supervision: AE, J-FC, CS, JEL, JLMF, BKM, RH, JC, LJC. Project administration: DG, LJC. Funding acquisition: LJC.
Funding Funding for this study was provided by The Leona M. and Harry B. Helmsley Charitable Trust (GCO ID 17-0378, L.J.C., J.C). Funding was also provided by the U24 DK062429, U01 DK062422 and the Sanford Grossman Charitable Trut (J.C.)
Competing interests J-FC reports receiving research grants from AbbVie, Janssen Pharmaceuticals, Prometheus, Takeda and Bristol Myers Squibb; receiving payment for lectures from AbbVie, and Takeda; receiving consulting fees from AbbVie, Amgen, AnaptysBio, Allergan, Arena Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Celgene Corporation, Celltrion, Eli Lilly, Ferring Pharmaceuticals, Galmed Research, Glaxo Smith Kline, Genentech (Roche), Janssen Pharmaceuticals, Kaleido Biosciences, Immunic, Invea, Iterative Scopes, Merck, Landos, Microba Life Science, Novartis, Otsuka Pharmaceutical, Pfizer, Protagonist Therapeutics, Prometheus, Sanofi, Seres, Takeda, Teva, TiGenix, Vifor; and hold stock options in Intestinal Biotech Development. JEL reports receiving research funding from Genentech and VectivBio, receiving consultant fees for Forte Biosciences, Incyte, Mesoblast, and Sanofi, and royalties from GVHD biomarker patent. LC reports receiving consultant fees for Orchard Therapeutics and ORGANOIDSCIENCES; receiving research grants from Bristol Myers Squibb; receiving payment for lectures from Ferring Pharmaceuticals and Takeda. JC reports receiving consultant fees for Orchard Therapeutics.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.