
Abstract
The landscape of the investigation and management of giant cell arteritis (GCA) is advancing. In this review we will outline the recent advances by searching the current English literature for relevant articles using key words of giant cell arteritis, temporal arteritis, Horton’s disease, investigation, and treatment. Delay in diagnosis, diagnostic uncertainty and glucocorticoid (GC) morbidity are among the highest concerns of clinicians and patients in this disease area. The positive news is that fast track pathways, imaging techniques and new therapies are emerging for routine management of GCA. Future directions for intervention in the treatment paradigm will be discussed.
摘要
巨细胞动脉炎 (GCA) 的诊疗发生了很大进展。在这篇综述中, 我们通过关键词搜索目前相关的英文文献, 以总结和回顾巨细胞动脉炎的最新进展。所使用的关键词为巨细胞动脉炎; 颞动脉炎; 霍顿病;观察;治疗等。在GCA疾病领域内, 临床医生和患者们最关注的问题是诊断延迟, 诊断不确定性和糖皮质激素 (GC) 的使用情况。好消息是诊疗快速通道的建立, 成像技术以及新治疗的日新月异已经应用于GCA的常规管理。在本文中我们也会探讨治疗、干预策略的新方向。
Similar content being viewed by others


Introduction
Giant cell arteritis (GCA) is a granulomatous medium and large-vessel vasculitis [1]. It is the most common form of systemic vasculitis, with an incidence of between 15 and 25 cases per 100,000 persons over 50 years of age [2]. The incidence of GCA increases with age, almost exclusively affecting people 50 years of age or older [3]. Women account for 65–75% of patients; the lifetime risk of GCA in women is 1% compared with 0.5% in men [3, 4]. It mainly affects Caucasians and has a higher incidence in Scandinavian countries and in populations of northern European descent [5]. It is rare in Asian and Black Caribbean/African populations [6, 7].
GCA remains a medical emergency because of the risk of sudden irreversible sight loss and stroke. It is a spectrum of phenotypically overlapping conditions including cranial GCA, extracranial GCA (otherwise termed large-vessel GCA, usually involving the aorta and its larger supra-aortic branches) and polymyalgia rheumatica (PMR) [5, 8]. The most commonly affected cranial arteries are the temporal, ophthalmic, posterior ciliary and vertebral arteries [9]. 22–83% of newly diagnosed GCA patients have imaging evidence of large-vessel GCA [10].
In clinical practice GCA can be multifaceted: this is due to the seriousness of the condition, diagnostic uncertainty and morbidity associated with treatment. Over the past decade there has been an increased awareness of GCA, leading to an increase in admissions for investigations for suspected GCA [11]. Thus, improved diagnostic pathways, specific tests and targeted management are imperative.
Pathophysiology of GCA
GCA mainly affects the medium and large arteries of the external cranial branches of the aorta. The pathological process occurring in GCA is summarised below [2, 3, 12, 13].
-
1.
Proceeding from an unknown trigger, there is abnormal maturation of vascular dendritic cells (DC) in the adventitia of the large-vessel walls. These activated DC recruit and activate cluster differentiation (CD) 4+ naïve T cells.
-
2.
Naïve CD4+ cells are activated and differentiate to T helper (Th) 1 cells, Th17 and T regulatory (Treg) cells.
-
3.
Macrophages within the tunica adventitia of the vessel wall produce IL-6 and IL-1β. Within the tunica media, macrophages secrete metalloproteinases, which degrade the internal elastic lamina and other connective tissue. Reactive oxygen species and secreted IL-6 contribute to inflammation, local vascular damage. Vascular damage and macrophage-derived growth factors such as VEGF and platelet derived growth factor cause intimal hyperplasia and subsequent vascular stenosis and occlusion.
-
4.
In some patients, IFN-γ promotes the differentiation and fusion of highly activated macrophages to form multinucleated giant cells. These giant cells also secrete cytokines and growth factors.
-
5.
The injured arterial cells respond to damage through dysfunctional repair. This leads to media thickening, luminal occlusion, ischaemia and eventually end organ damage.
Genetic influences in GCA
A genetic component to the development of GCA is supported by evidence of differential prevalence depending on the ethnicity, familial aggregation and multiple genetic associations. GCA has a strong association with the human leucocyte antigen (HLA) region, notably the HLA class II genes [14, 15], which suggests the role of the immune system in its pathophysiology. The number of identified loci remains low, and the reasons for this include the fact that GCA is not common within the general population making it hard to identify susceptibility signals [15].
Non-HLA associations have been associated with an increased risk of GCA, namely PLG (involved in the plasminogen system) and P4HA2 (collagen synthesis); both of these have an important role in neoangiogenesis [16]. Genes encoding proinflammatory cytokines can increase the patient’s risk of ischaemic complications, PMR and relapsing disease. The future potential for stratifying the disease, and providing targeted therapy has been postulated [14].
Role of infection in the development of GCA
A potential correlation between onset of GCA and infection is postulated: in analyses infections are found to be more common in GCA patients than non-GCA prior to their diagnosis [17, 18]. This leads to a number of theories regarding infection and GCA: namely that infections could be directly responsible for causing GCA from an unregulated inflammatory response, or that the pre-existing immune system alterations due to the genetic polymorphisms could increase both the risk of infection and development of vasculitis.
In support of the theory that infectious agents trigger GCA specific infectious agents have been found in temporal artery specimens of those diagnosed with GCA, with some then postulating them as a causative factor for the development of GCA. These include herpes simplex virus, chlamydia pneumonia, mycoplasma pneumonia, Epstein–Barr virus, parvovirus B19 [19] and varicella zoster virus (VZV) [20]. However, to date no clear conclusions can be drawn, as viruses such as VZV are ubiquitous and the data are currently conflicting.
Seasonal and geographic variation in GCA
Geo-environmental factors have been implicated in the development of GCA, and De Smit et al. not only investigated seasonal patterns of temporal artery biopsies (TAB) from Australia and New Zealand, but also summarised the literature [21]. They found that there was no statistically significant relationship between the season of presentation. On analysis of prior studies, they found results to be inconsistent with regard latitude, altitude, solar exposure, socioeconomic status and urban versus rural living again with little conclusion [21].
Disease associations
PMR is considered by some to be a variant of GCA, where overt vasculitis has either not commenced or is possibly prevented by an unknown inhibiting mechanism [5]. 40 to 60% of patients with GCA also have PMR, conversely 16–21% of PMR patients have GCA—perhaps resulting from sharing of genetic risk factors and pathogenesis [22]. GCA should be considered in PMR patients with marked constitutional symptoms and/or elevated acute phase reactants and where there is an inadequate response to GCs or relapse [23, 24]. PMR typically presents with bilateral shoulder pain and stiffness, which is abrupt in onset (usually reaching a peak within 2 weeks) and worse in the mornings [25]. Other symptoms suggestive of PMR include pelvic girdle aching for more than 2 weeks, morning stiffness for more than 45 min, functional impairment and constitutional symptoms [26]. However, exclusion of a wide range of imitating conditions is also required (e.g. other rheumatological illness such as osteoarthritis or rheumatoid arthritis, drug-induced, endocrine, infective and neoplastic conditions).
Other associations include a significant increased risk of peripheral arterial disease in GCA: the risk ratio among patients with GCA compared with controls was 1.88 (95% CI 1.04–3.41) [27]. Socioeconomic deprivation has been reported in GCA, independent of classic cardiovascular risk factors [28]; some consider this evidence that ischaemic complications result predominantly from diagnostic delay [29].
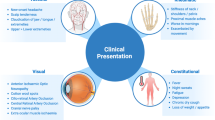
Clinical presentation—systemic features
Typical clinical features include new-onset headache, scalp tenderness, jaw claudication, fever, fatigue, malaise, anorexia, weight loss and polymyalgia [30]. In most cohorts, new-onset headache is typical in about two third of patients, whilst jaw claudication is present in under 50%. Jaw claudication is defined as pain and fatigue in the masseter muscles on protracted or vigorous chewing, which eases with rest. It should not be confused with TMJ dysfunction, dental or gum disease. Tongue claudication is less common, however if present, the likelihood of GCA increases [5]. Headache in GCA can be described as an ache, most commonly over the temporal arteries, but it can occur anywhere over the head. The region over the temporal artery may be sensitive to touch (termed allodynia). Temporal artery abnormalities such as beading (irregular contour), prominence, tenderness with an absent pulse are reported to increase the likelihood ratio for a positive TAB [31] (Fig. 1), although one third of biopsy proven GCA patients have normal temporal arteries on clinical examination.

Prominent, beaded, pulseless, tender temporal artery in a patient with biopsy proven giant cell arteritis. Abnormality detected by patient 3 weeks prior to new-onset temporary, then permanent double vision, followed by anterior ischaemic optic neuropathy and complete loss of vision to perception of light
Constitutional symptoms are present in up to 50% of patients with cranial GCA, including fever, fatigue, night sweats, anorexia and weight loss. These features can be dominant in 15% of patients at first presentation and 20% of patients when relapsing [5]. The fever associated with GCA is usually low grade, but with the potential to reach 39–40 °C. This is due to a strong systemic acute phase response [32]. Polymyalgic symptoms are the most common extracranial manifestation. Peripheral musculoskeletal manifestations are reported in GCA and PMR, including arthritis, swelling of the distal extremities with pitting oedema (more likely PMR than GCA), tenosynovitis and carpal tunnel syndrome [33], although their presence should alert the clinician to possible alternative diagnoses such as rheumatoid arthritis. Due to the wide ranging phenotype, clinicians require a high index of suspicion for GCA [29] because atypical presentations of GCA may delay diagnosis and lead to further complications [34].
The American College of Rheumatology (ACR) classification criteria were created in 1990 (Fig. 2). The presence of three or more of five criteria were associated with a sensitivity of 93.5% and a specificity of 91.2% for GCA when tested in a selected population of patients with vasculitis [35]. The criteria are often mistakenly used for diagnosis, where they function poorly. In the usual clinical setting such as a neuro-ophthalmology or rheumatology clinic they have low sensitivity and poor positive predictive value [36, 37]. They have incorrectly labelled GCA as a “headache disease” [24].

Understanding of the wider phenotype of GCA indicates that the ACR 1990 classification criteria require updating key clinical features of GCA such as constitutional and polymyalgic symptoms, major organ threatening features and newer imaging techniques such as ultrasound (US), CT, magnetic resonance imaging (MRI) and FDGPET should be included [5].
Other short and long-term complications of GCA include stenosis and large-vessel aneurysms, including thoracic and abdominal aortic aneurysm [38]. Clinically, these can present as intermittent limb claudication, back and chest pain. Rarer complications include stroke, tongue and scalp necrosis [3].
Clinical presentation—visual features
Visual symptoms can be temporary or permanent. Temporary double vision from cranial nerve palsy occurs in 6–27%. Amaurosis fugax, where there is complete black out of vision for seconds, is a concerning symptom and if disregarded permanent visual loss will occur. Previous visual loss rates, prior to the use of GCs, were reported between 30 and 60% [39]. Currently, vision loss occurs in up to 20% of patients with GCA, most commonly due to arteritic anterior ischaemic optic neuropathy, and in less than 5% due to choroidal infarction, central and branch retinal artery occlusions or posterior ischaemic optic neuropathy [1]. Risk factors for visual loss in GCA are older age, male gender, hypertension, a positive TAB and delayed initiation of GC [5, 40].
Securing a diagnosis
Making a diagnosis of GCA can be difficult due to the non-specific nature of some of the symptoms and a wide range of phenotypes [9]. Diagnosis can be challenging and requires a full history, thorough examination and a combination of investigations. It appears that investigations, along with clinician insight, produce the highest sensitivity and specificity [41].
Serological investigations
Evidence of an acute phase response is shown through raised erythrocyte sedimentation rate (ESR) or plasma viscosity (PV), and/or c-reactive protein (CRP) [42]. ESR was originally used in the diagnosis of GCA, but a combination of ESR and CRP is being used to give the highest sensitivity and specificity for a diagnosis of GCA. PV is not influenced by age, gender, time to analyse and haematocrit, therefore thought to be better than ESR if available [43]. Thrombocytosis, normocytic normochromic anaemia, with normal white blood cell count or mild leucocytosis can also be predictive of GCA [44]. Liver transaminases and alkaline phosphatase are often mildly elevated. A combination of tests have been found to be strong predictors of a positive diagnosis in those with suspected GCA [42].
Diagnostic uncertainty in GCA presenting to ophthalmology has increased due to the incorrect assumption of the term “occult GCA”. Occult GCA is defined in the ophthalmology literature as ocular signs without systemic symptoms, but with raised inflammatory markers [31, 45]. In a small percentage of patients, inflammatory markers may be normal [46]. Whilst inflammatory markers are usually highly elevated in GCA, lower CRP and/or ESR may be more common in those with visual loss. In 136 patients with biopsy proven GCA from one centre, the ESR and CRP values at diagnosis were significantly lower in patients with permanent visual loss [47]. In another multi-centre study of 32 patients with GCA, a low inflammatory response and the presence of transient cranial ischaemic events (such as amaurosis fugax and double vision) indicated a high risk of developing irreversible ischemic complications (odds ratio 5, 95% confidence interval 2.05–12.2) [48].
Imaging investigations
Cranial US
Vascular US is used to identify temporal artery oedema, stenosis and occlusion in GCA [41]. In ten studies (n = 696), the sensitivity has ranged from 55 to 100% and specificity from 78 to 100%. Typically the “halo sign” is observed, which is a non-compressible hypoechoic ring around an arterial lumen that represents an oedematous thickening of the arterial wall due to inflammation (Fig. 3) [49]. A meta-analysis published in 2010 found a sensitivity of 68% and a specificity of 91% for the unilateral halo sign. The bilateral halo sign was less sensitive, but had a specificity approaching 100% [50]. US of the axillary arteries as well as the temporal arteries increases sensitivity of the test [41, 51]. Careful clinical evaluation and assessment of the pre-test probability of diagnosis of GCA are still required, because the halo sign can rarely be found in other forms of vasculitis [52]. Further study is also needed to better define the confounding effects of atherosclerosis on temporal and axillary wall thickness [53]. Ultrasonography is cheaper than MRI; it is non-invasive, has rapid access and allows longitudinal scanning for disease activity; however it requires expertise [41]. The TABUL study investigated cranial US versus TAB and showed the sensitivity of TAB was 39%, which was inferior to US 54%; but the specificity of TAB was 100%, which was superior to US at 81%. Further analysis showed that when combined with clinical judgement at 2 weeks, the sensitivity of TAB increased to 91% and the sensitivity of US to 93%, with the specificity of 81% for TAB and 77% for US [41]. A cost saving of £485 was found in favour of US. The sensitivity and specificity of US in TABUL was lower than the most recent meta-analysis of US use in GCA, where pooled sensitivity was 77% and specificity 96% [54].

These are Temporal artery ultrasound images showing (a) longitudinal and (b) cross sectional images of a normal artery; (c) longitudinal and (d) cross section image of the non-compressible, hypoechoic “halo sign” (white asterisks)
Vascular US is more sensitive to GC therapy than TAB. In TABUL, US sensitivity was 64% if performed within one day of starting prednisolone and 47% if performed between 2 and 7 days [41]. Approximately half of positive US findings will be lost within 7 days of steroid treatment [43], although the halo sign may persist for months in some patients [55]. TABUL’s ultrasonographers had less scanning experience than others who have published in this field. Although training and access to US remain limitations for this technology, this is no reason not to try to develop local expertise [25]. The European League Against Rheumatism (EULAR) 2018 guidance recommends US as the first diagnostic test given adequate expertise and equipment, and if not available TAB [56].
18F-fluorodeoxyglucose positron emission tomography (18F-FDG-PET)
18F-FDG-PET is usually combined with low-dose computed tomography (CT) to detect inflamed vasculature seen in GCA, and this may have a role in assessing disease activity and extent of involvement (Fig. 4) [1]. Although large-vessel imaging is sensitive to GC therapy [57], uptake can persist despite treatment and absence of clinical symptoms in some patients [58]. There are no standardised criteria for 18F-FDG-PET in large-vessel vasculitis but a high proportion of clinicians agree with the benefits in GCA and diagnostic accuracy. It has shown a sensitivity of 77% and a specificity of 66% [59]. Whilst use in extracranial involvement of GCA is beneficial, it is less helpful for cranial GCA due to the proximity of the brain. Another limitation is in detecting vasculitis of the lower limb due to co-morbid atherosclerosis which can result in false positive results [60].

A 67-year-old Caucasian lady presented with headache, PMR, drenching night sweats, weight loss and CRP 120. 18FDGPET-CT fused images show 18FDG avid large-vessel GCA (white arrows) of the thoracic aorta in sagittal (a), coronal (b) and transverse view (c). Inflammatory activity extends into the subclavian and common carotid arteries (d)
Research in other imaging modalities
Research areas in imaging include super high-resolution MRI of the superficial and extracranial arteries, and transdermal optical coherence tomography (OCT) of the superficial temporal artery. A contrast-enhanced high-resolution MRI can demonstrate arterial wall thickening with peri-adventitial and mural contrast enhancement [1]. Neuroimaging is not usually required for patients with a typical presentation of GCA, but some patients may have already undergone the investigation previously [34]. Studies have shown MRI to have a sensitivity of 68–89% and a specificity of 73–97% [1]. Findings that can typically be seen in GCA include non-specific enhancement, enhancement of the optic nerve parenchyma, perineural sheath and optic chiasm. However, these findings are not exclusive to GCA, as they can suggest general orbital inflammatory disease, infiltrating or demyelinating disease. On MRI there can also be T2 hypersensitivity, gadolinium enhancement and diffusion weighted imaging restriction, all of which can be seen in GCA-related anterior ischemic optic neuropathy or posterior ischaemic optic neuropathy [34].
Transdermal OCT has been reported in a series of eight patients [61]; it demonstrated that transdermal OCT identified the superficial temporal artery. The advantages over TAB were reported to be similar to US and it is quick and easy to use. Currently more research would be required to validate the transdermal OCT utility in GCA.
Histopathological confirmation
A positive TAB gives a definitive diagnosis where a chronic granulomatous inflammation centred on internal elastic lamina is seen. The inflammation can be transmural or patchy comprising of mononuclear lymphohistiocytic cells and occasionally admixed neutrophils and eosinophils (Fig. 5a). Characteristic multinucleated giant cells (Fig. 5b) often accompany the inflammatory infiltrates, but these are not required for the diagnosis. Fragmentation and reduplication of the elastic laminae is usually present (Figs. 6 and 7b) [62]. Where timely non-invasive imaging is unavailable, an early TAB is recommended in all patients where there is suspicion of GCA [54, 63], as over time GCs will attenuate the histopathological features. Whilst a positive TAB is specific for GCA, it requires an invasive surgical procedure. Uncommon procedural risks include facial nerve injury, scalp ulcer/necrosis, discomfort and surgical wound complications. Stroke has been reported as an extremely rare complication during the surgery in one individual who later proved to have an asymptomatic carotid occlusion on the side of the surgery as the collateral flow had been passing through the biopsied artery [64]. There is a reported high false negative rate due to segmental involvement [29]. Since these skip lesions are common, a negative biopsy cannot completely rule out GCA. Deeper sections throughout the tissue blocks should be made if initial biopsy slides show only normal artery, and clinical history and examination should be revisited.

Key histopathological features of GCA. a H&E ×100, superficial temporal artery with marked intimal thickening. b Thick arrow head denotes inflammatory cells in the media and adventitia

Key histopathological feature of GCA with EHVG stain ×200 thick arrow head points to fragments of internal elastic laminae remaining and duplication

Key histopathological feature of healed arteritis. a Thick arrow head shows calcification at the intima media border and b an elastic stain (EHVG) highlights significant disruption of the internal elastic lamina with defects extending over 30–50% of the circumference of the artery
Another histologic pattern seen in TAB is the healed pattern, which shows no inflammation in the arterial wall but can demonstrate discontinuous elastic lamina, fibrosis of intima/ media and focal calcification (Fig. 7a). An elastic stain (EHVG) highlights significant disruption of the internal elastic lamina with defects extending over 30–50% of the circumference of the artery (Fig. 7b) [65]. Controversy in the literature exists around healed arteritis, and this should alert the clinician to re-examine the history and examination of the patient closely. In addition, atherosclerosis can be a confounding finding in TAB, as it causes intimal hyperplasia and fragmentation of the laminar. Thus, an experienced histopathologist has a critical role in helping to guide the clinician. The TABUL study [41] suggested significant variation between pathologists in the interpretation of TAB histology, so consideration should be made to discuss the pathology if the report does not fit the clinical picture.
The British Society for Rheumatology (BSR) recommends that TAB specimens should be no less than 1 cm and preferably more than 2 cm [66]. Post fixation shrinkage also needs to be taken into consideration [36]. Bilateral primary TAB is common in North America, and increases the yield, but is usually unnecessary [67].
The TABUL study found TAB had a 99% specificity and 39% sensitivity [41]. The sensitivity was much lower than others have reported. In one large cohort of patients with suspected GCA undergoing TAB, the main predictors of a positive biopsy were age at biopsy, referral from ophthalmology, jaw claudication/pain, visual symptoms and raised ESR [9]. In TABUL, TAB sensitivity dropped with GC therapy reported from 48 to 33% if performed within 3 days compared with 7 days [41]. The wider literature suggests that there is a 5–10% reduction in positive TAB rate for each week of glucocorticoid treatment [43].
The result of any diagnostic test should be interpreted in light of the pre-test probability. Pre-test probability is estimated by the treating clinician based on the history and examination findings, and all available laboratory results. Various algorithms or clinical prediction rules have been proposed to help with this, but clinical experience remains essential and is why, where possible, all patients with suspected GCA should be referred to a specialist [41].
Other assessments worth considering in GCA
Consider performing a screen for infection including dipstick urinalysis and chest X-ray (CXR); and a search for mimicking diseases with protein electrophoresis, thyroid function tests, and anti-cytoplasmic neutrophil antibodies. Baseline CXR, echocardiogram or large-vessel imaging are advocated by some centres to assess for large-vessel complications at baseline, in higher risk groups. Occasionally a contrast-enhanced MRI head and orbits may be indicated examining the anterior visual pathways where the cause of an optic neuropathy is not clear.
Determination of pre-existing comorbidities such as hypertension, diabetes mellitus, cataract, cardiovascular disease, peptic ulcer disease, osteoporosis and glaucoma is important as the treatment can initiate or worsen disease status (Fig. 8). Noting co-medication with non-steroidal anti-inflammatory drugs (NSAIDs) and anti-coagulants is important both for immediate and long-term care in GCA.

Percentage of new co-morbidities or adverse events recorded following a diagnosis of GCA cohort as adpated from Proven et al. [71]
Management of GCA
GCA is an emergency and many consider that it needs to be treated at the point of a suspected diagnosis due to the threat of sight loss. The principles of management of GCA are to protect vision, to limit other end organ damage and minimise GC toxicity and morbidity.
Fast track pathways
The mean diagnostic delay for those with cranial symptoms is 9 weeks and 17.6 weeks for patients presenting without cranial symptoms, due to a combination of multiple factors including delayed presentation, delayed clinical suspicion and delayed referral for specialist assessment and confirmatory diagnostic tests [68]. Public awareness of GCA is low and primary care doctors are faced with the often non-specific nature of many early symptoms of GCA and a high prevalence of similar symptoms in the general consulting population [68]. If GCs are started, but specialist referral is delayed, then the potential for a secure diagnosis is reduced, the clinical signs may have resolved and the diagnostic tests are less likely to be positive with increasing time.
To address these challenges, in certain centres fast track pathways have been established, providing widely advertised, rapid access to specialist clinical assessment, diagnostic evaluation including vascular US or other diagnostic tests and immediate therapy. Such pathways have been shown to reduce sight loss and have led to significant cost savings due to reduction of inpatient care [41] and reduction in TAB rate [41, 52]. In one fast track centre, sight loss was reduced from 37 to 9% [40], in another from 27 to 8% [55]. In a third centre, although visual disturbance was equal in the historical and fast track groups, risk of permanent blindness reduced by 88% in the fast track group [69].
Immediate treatment
Immediate GCs are the treatment of choice for patients with suspected GCA, however there is uncertainty regarding the optimum dose [1]. Patients who present to the ophthalmology community with cranial ischaemic symptoms and/or ophthalmic symptoms are traditionally offered high dose pulse therapy of GC with 0.5–1.0 g methylprednisolone for 3–5 days [63, 66]. This reduces the cumulative dose of oral GC [1], but there is no evidence that this reduces rates of sight loss. The current BSR and EULAR guidelines suggest immediate treatment of GCA using 1 mg/kg GC (up to a maximum of 60 mg/day) or 40–60 mg/day equivalent [63]. This is to reduce the risk of ischaemic complications, especially to prevent vision loss [63, 66]. For those without cranial ischaemic symptoms, an initial dose of 40 mg/day is deemed adequate.
Maintenance therapy
Tapering regimens are empirical. The BSR guidance recommends a GC taper over about 1–2 years, although this may need to be adjusted according to response [66].
Suggested tapering regime by BSR [66] |
• 40–60 mg prednisolone until symptoms and lab results are normal (2–4 weeks) |
• Reduce dose by 10 mg every 2 weeks to 20 mg |
• Reduce dose by 2.5 mg every 2–4 weeks to 10 mg |
• Reduce dose by 1 mg every 1–2 months if there is no relapse |
Observational cohort studies report relapses in 34–62% of patients, requiring an increase in GC dose, and/or slowing of the tapering regimen [24]. Patients should be warned to expect treatment for about 2 years, whilst most patients are off GCs at 4–5 years. A small percentage of patients need GCs indefinitely [43]. Those who are at high risk of more prolonged GC therapy or relapse include women, those with peripheral arthritis, high initial inflammatory markers and those with evidence of large-vessel involvement [1, 70]. Due to the protracted GC burden, there may be significant adverse effects [71].
Six percent of patients receiving steroid therapy develop serious neuropsychiatric complications [72]. This effect is dose dependent, with the incidence rapidly rising once at 40 mg/day of steroid [73]. Other adverse effects include the development of a cushingoid appearance, weight gain and skin atrophy. There are also comorbidities that can be exacerbated by GC therapy, including diabetes, glaucoma and osteoporosis [1].
Adjunctive therapy in GCA
One of the key areas for development in GCA has been to bridge the unmet need for adjunctive therapy. GCA patients typically exceed a cumulative dose of 5000 mg prednisolone over several years with up to 85% experiencing GC related side effects [71]. In the UK alone over 33% had a reported cumulative dose over 10,000 mg [74]: the adverse event hazard ratio rising by 3% for every 1000 mg increase [74]. In addition, those with disease refractory to GCs (Table 1) and those intolerant or with significant adverse effects from GCs (Fig. 8) require adjunctive treatment. Disapointingly a number of agents have been unsuccessful for adjunctive treatment in GCA (Table 2).
Methotrexate
Methotrexate has been tested as a steroid-sparing agent and in refractory GCA [75,76,77]. Currently EULAR recommends considering methotrexate in every patient diagnosed with GCA, whereas BSR recommends treatment for patients with refractory disease [63, 66] based on evidence from three small RCTs [75,76,77]. A meta-analysis of these trials displays only modest efficacy of 7.5–15 mg methotrexate once per week as an adjunct in GCA [78]. Doses of 15–25 mg methotrexate weekly, which are conventionally used, have not been formally tested in GCA. The most common adverse effects include gastrointestinal upset, mouth ulcers, temporary hair loss, elevated liver enzymes, leukopenia and teratogenicity [1].
Aspirin
Aspirin is an oral anti-platelet drug, which reduces platelet aggregation, thereby inhibiting thrombosis formation [29]. It also has a wide range of effects on the immune system, inducing tolerance in DCs, inducing Treg and suppressing the transcription of IFN-γ [79, 80]. Since GCs only have marginal effects on IFN-γ, aspirin should theoretically complement their effect in GCA [29]. Due to its antithrombotic action, a small case series of GCA patients have supported its use to reduce the risk of ischaemic events [81]. However, there is a lack of adequate evidence supporting the use of aspirin in GCA, so the clinician must weigh up the potential benefits over potential morbidity [29]. Adverse effects of aspirin include gastrointestinal and cerebral haemorrhage, which will be exacerbated by co-prescription with GC.
Cyclophosphamide
In a case series of 35 patients with refractory GCA, 90.3% (n = 28) responded to cyclophosphamide with improved disease activity and sustained reduction in prednisolone dose [82]. A literature review of GCA patients treated with cyclophospahmide [83] showed 84% (n = 88) were responsive, alongside other immunosuppressive agents as part of maintenance therapy. However, the rate of side effects was high (33%), and the positive result likely to be affected by reporting bias. The lack of prospective or RCT data and the side effects of cyclophosphamide, especially in this older group of patients, including life threatening infection, limit its use in GCA.
Mycophenolate
There has been one small case series (n = 3) suggesting potential of using mycophenolate in GCA [84].
Leflunomide
This inhibits DC maturation and decreases production of IL-6, tumour necrosis factor (TNF), IL-12 and IL-17. It has been shown to have efficacy in Takayasu arteritis and granulomatosis with polyangiitis [85] and is widely used in both psoriatic and rheumatoid arthritis. The most common side effects include diarrhoea, raised blood pressure, elevated liver enzymes and leukopenia. There are case series highlighting its use as a steroid-sparing agent in large-vessel vasculitis [85, 86]. Adizei et al. [86] showed 22 out of 23 difficult-to-treat GCA patients had either a partial or complete response, as well as a reduced cumulative GC dose. Diamantopoulos et al. [85] showed good responses with 11 refractory GCA patients. Recently an open label study comparing leflunomide as adjunctive therapy to standard care and GCs alone found that three quarters of the leflunomide group had reduced relapse rate and decreased cumulative doses of GCs. Leflunomide needs further high-level evidence to support its use in GCA [87].
Anti-TNF Therapy
TAB specimens show TNF in abundance in patients with GCA [88]. The exact role of TNF in the pathogenesis of GCA is still unclear and the role of anti-TNF therapy has not been studied with large sample sizes. Anti-TNF therapies have been considered for use in refractory cases of GCA or corticosteroid dependence, but are not routinely recommended (Table 1) [89].
The new era for GCA: targeted treatment
Tocilizumab (TCZ)
TCZ is a recombinant humanised anti-IL-6 receptor antibody. This competitively inhibits the binding of IL-6 to the membrane bound and soluble IL-6 receptors. IL-6 itself stimulates the release of ESR and CRP from hepatocytes, and promotes the transition from acute to chronic inflammation. Its overproduction contributes to the pathogenesis of GCA [90]. Using drugs that inhibit IL-6 should thereby halt this inflammatory process.
The GiACTA trial [91] investigated the efficacy of 162 mg subcutaneous TCZ with a 26-week prednisolone taper, either weekly or every other week. They found that there was a significantly higher rate of sustained remission in the TCZ arms than prednisolone arms (14% in the 26-week prednisolone taper, 18% in the 52-week prednisolone taper 56% in the weekly TCZ arm and 53% in the alternate week TCZ arm). The time to first flare was also significantly longer in the TCZ arms. TCZ reduced the cumulative median prednisolone dose by over 40% and perhaps as a result, reported serious adverse events were lower in the TCZ arms.
TCZ has been widely used in rheumatoid arthritis for about 10 years and a Cochrane review (n = 3334) [92] showed that TCZ was generally well tolerated. However there is a statistically significant association with changes in liver enzymes and total triglycerides and cholesterol [93]. Other adverse drug reactions included gastrointestinal haemorrhage and perforation, infection, headache and hypertension [94]. There is also a risk of injection site reactions when the drug is administered subcutaneously [95]. The risk of tuberculosis reactivation, malignancy and hepatitis was not found to be significantly elevated [93]. A past medical history of diverticulitis, may be a contraindication for use in GCA, because of the risk of perforation, especially if TCZ is to be used in conjunction with NSAIDs, GCs or methotrexate [13]. However, at the present time, in the absence of any other effective and approved GC-sparing agent in GCA, the risk of perforation must be balanced against the risk of ongoing active disease and high dose GC use. Studies have shown that there are no differences in the safety profile of TCZ in regards to sex, age, concurrent treatment with NSAIDs, methotrexate and GCs or mild renal impairment [96]. The safety of TCZ in patients with moderate to severe renal impairment, hepatic impairment or in pregnancy have not yet been studied. Currently, there is an extension study (ClinicalTrials.gov Identifier: NCT03202368) investigating the long-term safety of subcutaneous TCZ in patients with GCA who have flare or persisting disease activity.
The National Institute for Health and Care Excellence and NHS England have approved TCZ for restricted use in GCA [97]. It is currently limited to refractory and relapsing disease (Table 1), in those who have not received TCZ previously, and for a maximum of one calendar year of treatment. In the GiACTA trial [71], 17% of the overall cohort were diagnosed with refractory GCA. Within the cohort with refractory GCA, there was a higher prevalence of depression (16 versus 4%), and osteopenia/osteoporosis (33 versus 23%) compared with those without refractory GCA.
Anti-IL-1 therapy
Ly et al. [98] reported the efficacy of anakinra in three patients with refractory GCA, in comparison to conventional treatments. The GiAnT trial (ClinicalTrials.gov Identifier: NCT02902731) is currently in phase 3 looking at 70 patients who will be randomised to two arms: prednisolone plus placebo or prednisolone and 100 mg/day anakinra by subcutaneous injection. The study is expected to be completed by March 2022.
Anti-T-cell therapy
Langford et al. 2017 [99] (n = 49) supported the use of abatacept as it showed a significantly higher rate of relapse-free survival to standard GC therapy and it was not associated with a higher rate of toxicity. A clinical trial (ClinicalTrial.gov Identifier: 00556439) is currently underway looking at abatacept for treating adults with GCA and Takayasu’s arteritis.
Conclusion
GCA remains a disease that causes concern for clinicians and patients either in diagnosis or in suspected relapse. Wider access and validation of imaging techniques, not only for diagnosis but longitudinal monitoring, is underway. We are now embarking on the era of targeted treatment for GCA, with many avenues to pursue. Patients will benefit from fast track pathways where close partnership with other teams that care for those with GCA including rheumatology, medicine, neurology and primary care, will facilitate early diagnosis and improve long-term management.
Summary
What was known before
-
TAB was the “gold standard” for diagnosis of GCA.
-
Serum IL-6 was found to be elevated in those with active GCA.
-
The only effective treatment was with high dose GCs, which have a significant cumulative morbidity in this population.
What this study adds
-
Tests including non-invasive imaging in conjunction with clinical expertise are the current gold standard for diagnosis of GCA.
-
Fast track pathways reduce GCA-related morbidity, such as visual loss; reduce the potential for over treatment in those with suspected in GCA and are cost effective.
-
Tocilizumab, an IL-6 inhibitor, is licenced for treatment of all GCA patients. In Scotland, Wales and Northern Ireland it is funded for all GCA patients. In England it funded for refractory GCA for up to 1 year of treatment.
References
Buttgereit F, Dejaco C, Matteson E, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA. 2016;315:2442–58.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349:160–9.
Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. 2014;371:50–7.
Crowson CS, Matteson EL, Myasoedova E, Michet CJ, Ernste FC, Warrington KJ, et al. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011;63:633–9.
Dejaco C, Duftner C, Buttgereit F, Matteson EL, Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatology. 2017;56:506–15.
Smith C, Fidler W, Pinals R. The epidemiology of giant cell arteritis: report of a ten-year study in Shelby County, Tennessee. Arthritis Rheum. 1983;26:1214–9.
Wigley R, Zhang N, Zeng Q, Shi C, Hu D, Couchman K, et al. Rheumatic diseases in China: ILAR-China study comparing the prevalence of rheumatic symptoms in northern and southern rural populations. J Rheumatol. 1994;21:1484–90.
Koster M, Matteson E, Warrington KJ . Large-vessel GCA: diagnosis, monitoring and management. Rheumatology. 2018;57:32–42.
Gajree S, Borooah S, Dhillon N, Goudie C, Smith C, Aspinall P. Temporal artery biopsies in South East Scotland: a five year review. J R Coll Physicians Edinb. 2017;47:124–8.
Kermani T, Warrington KJ, Crowson CS, Ytterberg SR, Hunder GG, Gabriel SE, et al. Large-vessel involvement in giant cell arteritis: a population-based cohort study of the incidence-trends and prognosis. Ann Rheum Dis. 2013;72:1989–94.
Mollan SP, Begaj I, Mackie S, O’Sullivan EP, Denniston AK. Increase in admissions related to giant cell arteritis and polymyalgia rheumatica in the UK, 2002-13, without a decrease in associated sight loss: potential implications for service provision. Rheumatology. 2015;54:375–7.
Al-Mousawi AZ, Gurney SP, Lorenzi AR, Pohl U, Dayan M, Mollan SP. Reviewing the pathophysiology behind the advances in the management of Giant Cell Arteritis. Ophthalmol Ther. 2019;8:177–93. in progress
Mollan SP, Horsburgh J. Profile of tocilizumab and its potential in the treatment of giant cell arteritis. Eye Brain. 2018;10:1–11.
Carmona F, Martín J, Gonzalez-Gay MA. New insights into the pathogenesis of giant cell arteritis and hopes for the clinic. Expert Rev Clin Immunol. 2016;12:57–66.
Ortiz-Fernández L, Carmona FD, López-Mejías R, et al. Cross-phenotype analysis of Immunochip data identifies KDM4C as a relevant locus for the development of systemic vasculitis. Ann Rheum Dis. 2018;77:589–95.
Carmona F, Vaglio A, Mackie S, Hernández-Rodríguez J, Monach PA, Castañeda S, et al. A genome-wide association study identifies risk alleles in plasminogen and P4HA2 associated with giant cell arteritis. Am J Hum Genet. 2017;100:64–74.
Russo M, Waxman J, Abdo A, Serebro L. Correlation between infection and the onset of the giant-cell (temporal) arteritis syndrome - a trigger mechanism. Arthritis Rheum. 1995;38:374–80.
Brault C, Riis AH, Mor A, Duhaut P, Thomsen RW. Does low risk of infections as a marker of effective immunity predict increased risk of subsequent giant cell arteritis or polymyalgia rheumatica? A Danish population-based case-control study. Clin Epidemiol. 2018;10:1533–43.
Álvarez-Lafuente R, Fernández-Gutiérrez B, Jover JA, Júdez E, Loza E, Clemente D, et al. Human parvovirus B19, varicella zoster virus, and human herpes virus 6 in temporal artery biopsy specimens of patients with giant cell arteritis: analysis with quantitative real time polymerase chain reaction. Ann Rheum Dis. 2005;64:780–2.
Procop GW, Eng C, Clifford A, et al. Varicella zoster virus and large vessel vasculitis, the absence of an association. Pathog Immun. 2017;2:228–38.
De Smit E, Clarke L, Sanfilippo PG, et al. Geo-epidemiology of temporal artery biopsy-positive giant cell arteritis in Australia and New Zealand: is there a seasonal influence? RMD Open. 2017;3:e000531 https://doi.org/10.1136/rmdopen-2017-000531.
Gonzalez-Gay MA. Giant cell arteritis and polymyalgia rheumatica: two different but often overlapping conditions. Semin Arthritis Rheum. 2018;33:289–93.
Dejaco C, Singh YP, Perel P, Hutchings A, Camellino D, Mackie S, et al. 2015 recommendations for the management of polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Arthritis Rheumatol. 2015;67:2569–80.
Dejaco C, Brouwer E, Mason J, Buttgereit F, Matteson EL, Dasgupta B. Giant cell arteritis and polymyalgia rheumatica: current challenges and opportunities. Nat Rev Rheumatol. 2017;13:578–92.
Quick V, Kirwan J. Our approach to the diagnosis and treatment of polymyalgia rheumatica and giant cell (temporal) arteritis. J R Coll Physicians Edinb. 2012;42:341–9.
González-Gay MA, Matteson EL, Castañeda S. Polymyalgia rheumatica. Lancet. 2018;390:1700–12.
Ungprasert P, Thongprayoon C, Srivali N, Cheungpasitporn W. Peripheral arterial disease in patients with giant cell arteritis: a meta-analysis. Int J Rheum Dis. 2016;19:819–25.
Mackie S, Dasgupta B, Hordon L, Gough A, Green M, Hollywood J, et al. Ischaemic manifestations in giant cell arteritis are associated with area level socio- economic deprivation, but not cardiovascular risk factors. Rheumatology. 2011;50:2014–22.
Mollan S, Sharrack N, Burdon M, Denniston A. Aspirin as adjunctive treatment for giant cell arteritis. Cochrane Database Syst Rev. 2014;8:CD010453.
Hunder G. Clinical features of GCA/PMR. Clin Exp Rheumatol. 2000;18:6–8.
Smetana G, Shmerling R. Does this patient have temporal arteritis? JAMA. 2002;287:92–101.
Salvarani C, Cantini F, Boiardi L, Hunder G. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med. 2002;347:261–71.
Salvarani C, Hunder G. Musculoskeletal manifestations in a population‐based cohort of patients with giant cell arteritis. Arthritis Rheumatol. 1999;42:1259–66.
D’Souza NM, Morgan ML, Almarzouqi SJ, Lee AG. Magnetic resonance imaging findings in giant cell arteritis. Eye. 2016;30:1–5.
Hunder GG, Bloch D, Michel B, Stevens M, Arend W, Calabrese L, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33:1122–8.
Murchison A, Gilbert M, Bilyk J, Eagle R, Pueyo V, Sergott R, et al. Validity of the American College of Rheumatology Criteria for the diagnosis of giant cell arteritis. Am J Ophthalmol. 2012;154:722–9.
Rao J, Allen N, Pincus T. Limitations of the 1990 ACR rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med. 1998;129:345–52.
Evans J, O’Fallon W, Hunder G. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis: a population-based study. Ann Intern Med. 1995;122:502–7.
Birkhead N, Wagener H, Shick R. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc. 1957;163:821–7.
Patil P, Williams M, Maw W, Achilleos K, Elsideeg S, Dejaco C, et al. Fast track pathway reduces sight loss in giant cell arteritis: results of a longitudinal observational cohort study. Clin Exp Rheumatol. 2015;33:103–6.
Luqmani R, Lee E, Singh S, Gillett M, Schmidt W, Bradburn M, et al. The role of ultrasound compared to biopsy of temporal arteries in the diagnosis and treatment of giant cell arteritis (TABUL): a diagnostic accuracy and cost-effectiveness study. Heal Technol Assess. 2016;20:1–238.
Ing E, Lahaie L, Toren A, Ing R, Chen J, Arora N, et al. Multivariable prediction model for suspected giant cell arteritis: development and validation. Clin Ophthalmol. 2017;11:2031–42.
Quick V, Kirwan J. In: Younger DS, editor. The vasculitidies, general considerations and systemic vasculitis Vol.1. New York: Nova Science; 2015. Chapter 17. p. 311–336.
Foroozan R, Danesh-Meyer H, Savino P, Gamble G, Mekari-Sabbagh O, Sergott R. Thrombocytosis in patients with biopsy-proven giant cell arteritis. Ophthalmology. 2002;109:1267–71.
Cullen J. Occult temporal arteritis. Trans Ophthalmol Soc UK. 1963;83:725–36.
Yu-Wai-Man P, Dayan M. Giant cell arteritis with normal inflammatory markers. Acta Ophthalmol Scand. 2007;85:460.
Salvarani C, Cimino L, Macchioni P, Consonni D, Cantini F, Bajocchi G, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum. 2005;53:293–7.
Cid M, Font C, Oristrell J, de la Sierra A, Coll-Vinent B, Lopez-Soto A, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. 1998;41:26–32.
Schafer V, Chrysidis S, Dejaco C, Duftner C, Lagnocco A, Bruyn G, et al. Assessing vasculitis in giant cell arteritis by ultrasound: results of OMERACT patient-based reliability exercises. J Rheumatol. 2018;45:1289–95.
Arida A, Kyprianou M, Kanakis M, Sfikakis PP. The diagnostic value of ultrasonography-derived edema of the temporal artery wall in giant cell arteritis: a second meta-analysis. BMC Musculoskelet Disord. 2010;11:44.
Schmidt W, Seifert A, Gromnica-Ihle E, Krause A, Natusch A. Ultrasound of proximal upper extremity arteries to increase the diagnostic yield in large-vessel giant cell arteritis. Rheumatology. 2008;47:96–101.
Monti S, Floris A, Ponte C, Schmidt W, Diamantopoulos A, Pereira C, et al. The use of ultrasound to assess giant cell arteritis: review of the current evidence and practical guide for the rheumatologist. Rheumatology. 2018;57:227–35.
Sammel A, Hamilton D, Omari A, McGrath M. Comment on: the proposed role of ultrasound in the management of giant cell arteritis in routine clinical practice. Rheumatology. 2018;57:1124–5.
Duftner C, Dejaco C, Sepriano A, Falzon L, Schmidt W, Ramiro S. Imaging in diagnosis, outcome prediction and monitoring of large vessel vasculitis: a systematic literature review and meta-analysis informing the EULAR recommendations. RMD Open. 2018;4:e000612.
Schmidt W. Ultrasound in the diagnosis and management of giant cell arteritis. Rheumatology. 2018;57:22–31.
Dejaco C, Ramiro S, Duftner C, Besson F, Bley T, Blockmans D, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis. 2018;77:636–43.
Hauenstein C, Reinhard M, Geiger J, Markl M, Hetzel A, Treszl A, et al. Effects of early corticosteroid treatment on magnetic resonance imaging and ultrasonography findings in giant cell arteritis. Rheumatology. 2012;51:1999–2003.
Blockmans D, de Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum. 2006;55:131–7.
Blockmans D, Stroobants S, Maes A, Mortelmans L. Positron emission tomography in giant cell arteritis and polymyalgia rheumatica: evidence for inflammation of the aortic arch. Am J Med. 2018;108:246–9.
Förster S, Tato F, Weiss M, Czihal M, Rominger A, Bartenstein P, et al. Patterns of extracranial involvement in newly diagnosed giant cell arteritis assessed by physical examination, colour coded duplex sonography and FDG-PET. Vasa. 2011;40:219–27.
Mollan S, Keane P, Denniston A. The use of transdermal optical coherence tomography to image the superficial temporal arteries. Eye. 2017;31:157–60.
Nesher G. The diagnosis and classification of giant cell arteritis. J Autoimmun. 2014;48–49:73–5.
Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2009;68:318–23.
Schlezinger NS, Schatz NJ. Giant cell arteritis (temporal arteritis). Trans Am Neurol Assoc. 1971;96:12–15.
Allsop C, Gallagher P. Temporal artery biopsy in giant-cell arteritis. A reappraisal. Am J Surg Pathol. 1981;5:317–23.
Dasgupta B, Borg FA, Hassan N, Alexander L, Barraclough K, Bourke B, et al. Guidelines BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology. 2010;49:1594–7.
Boyev L, Miller N, Green W. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol. 1999;128:211–5.
Prior J, Ranjbar H, Belcher J, Mackie S, Helliwell T. Diagnostic delay for giant cell arteritis - a systematic review and meta-analysis. BMC Med. 2017;15:120.
Diamantopoulos A, Haugeberg G, Lindland A, Myklebust G. The fast-track ultrasound clinic for early diagnosis of giant cell arteritis significantly reduces permanent visual impairment: towards a more effective strategy to improve clinical outcome in giant cell arteritis? Rheumatology. 2016;55:66–70.
Blockmans S, Coudyzer W, Vanderschueren S, Stroobants S, Loeckx D, Heye S, et al. Relationship between fluorodeoxyglucose uptake in the large vessels and late aortic diameter in giant cell arteritis. Rheumatology. 2008;47:1179–84.
Proven A, Gabriel SE, Orces C, O'Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum. 2003;49:703–8.
Dubovsky A, Arvikar S, Stern T, Axelrod L. The neuropsychiatric complications of glucocorticoid use: steroid psychosis revisited. Psychosomatics 2012;53:103–15.
West S, Kenedi C. Strategies to prevent the neuropsychiatric side-effects of corticosteroids: a case report and review of the literature. Curr Opin Organ Transpl. 2014;19:201–8.
Broder M, Sarsour K, Chang E, Collinson N, Tuckwell K, Napalkov P, et al. Corticosteroid-related adverse events in patients with giant cell arteritis: a claims-based analysis. Semin Arthritis Rheum. 2016;46:246–52.
Spiera R, Mitnick H, Kupersmith M, Richmond M, Spiera H, Peterson M, et al. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol. 2001;19:495–501.
Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46:1309–18.
Jover JA, Hernández-García C, Morado I, Vargas E, Banares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2001;134:106–14.
Mahr A, Jover J, Spiera R, Hernández-García C, Fernández-Gutiérrez B, Lavalley M, et al. Adjunctive methotrexate for treatment of giant cell arteritis. Arthritis Rheum. 2007;56:2789–97.
Hussain M, Javeed A, Ashraf M, Zhao Y, Mukhtar MM, Rehman MU. Aspirin and immune system. Int Immunopharmacol. 2012;12:10–20.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum. 2002;46:457–66.
Rüegg S, Engelte S, Jeanneret C, Hetzel A, Probst A, Steck A, et al. Bilateral vertebral artery occlusion resulting from giant cell arteritis: report of 3 cases and review of the literature. Medicine. 2003;82:1–12.
Loock J, Henes J, Kotter I, Witte T, Lamprecht P, Schirmer M, et al. Treatment of refractory giant cell arteritis with cyclophosphamide: a retrospective analysis of 35 patients from three centres. Clin Exp Rheumatol. 2012;30 1 suppl 70:70–6.
de Boysson H, Boutemy J, Creveuil C, Ollivier Y, Letellier P, Pagnoux C, et al. Is there a place for cyclophosphamide in the treatment of giant-cell arteritis? A case series and systematic review. Semin Arthritis Rheum. 2013;43:105.
Sciascia S, Piras D, Baldovino S, Russo A, Naretto C, Rossi D, et al. Mycophenolate mofetil as steroid-sparing treatment for elderly patients with giant cell arteritis: report of three cases. Aging Clin Exp Res. 2012;24:273–7.
Diamantopoulos AP, Hetland H, Myklebust G. Leflunomide as a corticosteroid-sparing agent in giant cell arteritis (GCA) and polymyalgia rheumatica (PMR): A consecutive case series. Biomed Res Int. 2013;2013:120638.
Adizie T, Christidis D, Dharmapaliah C, Borg F, Dasgupta B. Efficacy and tolerability of leflunomide in difficult‐to‐treat polymyalgia rheumatica and giant cell arteritis: a case series. Int J Clin Pr. 2012;66:906–9.
Hočevar A, Ješe R, Rotar Z, Tomšič M. Does leflunomide have a role in giant cell arteritis? An open-label study. Clin Rheumatol. 2018;38:1–6.
Martínez-Taboada VM, Rodríguez-Valverde V, Carreño L, López-Longo J, Figueroa M, Belzunegui J, et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis. 2008;67:625–30.
Seror R, Baron G, Hachulla E, Debandt M, Larroche C, Puéchal X, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis. 2014;73:2074–81.
Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:18460–5.
Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017;377:317–28.
Singh J, Beg S, Lopez-Olivo M. Tocilizumab for rheumatoid arthritis: a cochrane systematic review. J Rheumatol. 2011;38:10–20.
Genovese M, McKay J, Nasonov E, Mysler E, da Silva N, Alecock E, et al. Interleukin‐6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease‐modifying antirheumatic drugs: the tocilizumab in combination with traditional disease‐modifying antirheumatic drug the. Arthritis Rheum. 2008;58:2968–80.
Jones G, Sebba A, Gu J, Lowenstein MB, Calvo A, Gomez-Reino JJ, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69:88–96.
Burmester GR, Rubbert-Roth A, Cantagrel A, Hall S, Leszczynski P, Feldman D, et al. Efficacy and safety of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional DMARDs in patients with RA at week 97 (SUMMACTA). Ann Rheum Dis. 2016;75:68 LP–74.
Sheppard M, Laskou F, Stapleton PP, Hadavi S, Dasgupta B. Tocilizumab (Actemra). Hum Vaccin Immunother 2017;13:1972–88.
National Institute for Health and Care Excellence. Tocilizumab for treating giant cell arteritis. NICE technology appraisal guidance TA518. 2018. https://www.nice.org.uk/guidance/ta518.
Ly KH, Stirnemann JÔ, Liozon E, Michel M, Fain O, Fauchais AL. Interleukin-1 blockade in refractory giant cell arteritis. Jt Bone Spine 2014;81:76–8.
Langford CA, Cuthbertson D, Ytterberg SR, Khalidi N, Monach PA, Carette S, et al. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol. 2017;69:837–45.
Vodopivec I, Rizzo J. Ophthalmic manifestations of giant cell arteritis. Rheumatology. 2018;57:63–72.
Calvo-Romero J. Giant cell arteritis. Postgr Med J. 2003;79:511–5.
Coath F, Gillbert K, Griffiths B, Hall F, Kay L, Lanyon P, et al. Giant cell arteritis: new concepts, treatments and the unmet need that remains. Rheumatology. 2018;58:1316.
De Silva M, Hazleman BL. Azathioprine in giant cell arteritis/polymyalgia rheumatica: a double-blind study. Ann Rheum Dis. 1986;45:136–8.
Schaufelberger C, Möllby H, Uddhammar A, Bratt J, Nordborg E. No additional steroid‐sparing effect of cyclosporine A in giant cell arteritis. Scand J Rheumatol. 2006;35:327–9.
Liozon F, Vidal E, Barrier J. Dapsone in giant cell arteritis treatment. Eur J InternMed. 1993;4:207–14.
Hoffman GS, Cid MC, Rendt-Zagar K, Merkel P, Weyand C, Stone J, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621–30.
Funding
AJS is funded by an NIHR Clinician Scientist Fellowship (NIHR-CS-011-028) and by the Medical Research Council, UK (MR/K015184/1).
Author information
Authors and Affiliations
Contributions
HSL: literature review, and drafting of the manuscript and figures. VQ: Rheumatology Expert in GCA; drafting and critical review of the manuscript. Provision of the imaging figures. AJS: Neurology Expert in headache; critical review of the manuscript. SN: Neurohistopathologist; critical review of the manuscript and provision of the histopathology images. SPM: concept and design of the review; critical review of the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
SPM has undertaken consultancy work (speaker and advisory board fees) from Roche. VQ has undertaken consultancy work (speaker and advisory board fees) for Roche. Other authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lyons, H.S., Quick, V., Sinclair, A.J. et al. A new era for giant cell arteritis. Eye 34, 1013–1026 (2020). https://doi.org/10.1038/s41433-019-0608-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-019-0608-7
This article is cited by
-
Causal genes identification of giant cell arteritis in CD4+ Memory t cells: an integration of multi-omics and expression quantitative trait locus analysis
Inflammation Research (2025)
-
Step forward towards treat-to-target management of giant cell arteritis: patients stratification aiming to targeted remission – updated guidelines
Egyptian Rheumatology and Rehabilitation (2024)
-
Ischämien der Zunge und der Augenmuskeln als „mimic“ eines Hirnstamminfarkts bei 73-jähriger Patientin mit erhöhten Entzündungswerten
DGNeurologie (2024)
-
Temporal arteritis: iconodiagnosis in two 16th-century works of art, and a brief review of the medical and artistic literature
Neurological Sciences (2024)
-
Giant cell arteritis: reviewing the advancing diagnostics and management
Eye (2023)
